Wetenschap
Een game changer:metagenomische clustering mogelijk gemaakt door supercomputers
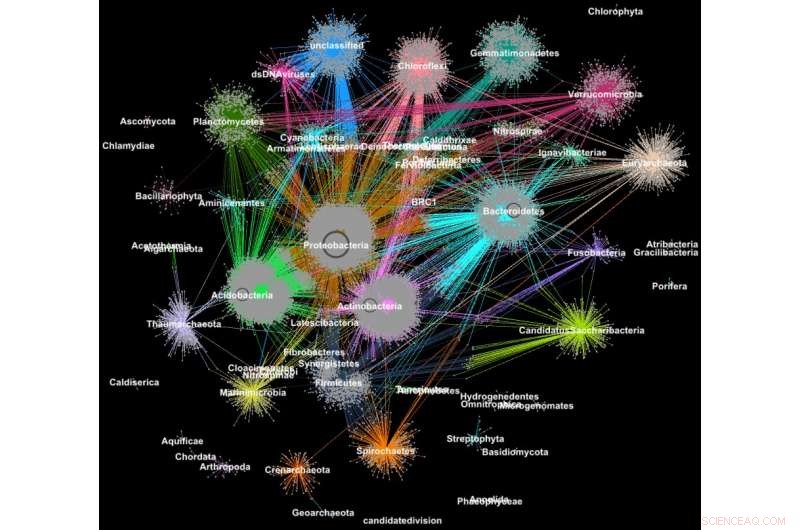
Eiwitten van metanomen zijn geclusterd in families volgens hun taxonomische classificatie. Krediet:Georgios Pavlopoulos en Nikos Kyrpides, JGI/Berkeley Lab
Wist u dat de tools die worden gebruikt voor het analyseren van relaties tussen gebruikers van sociale netwerken of het rangschikken van webpagina's ook zeer waardevol kunnen zijn om grote wetenschappelijke gegevens te begrijpen? Op een sociaal netwerk als Facebook, elke gebruiker (persoon of organisatie) wordt weergegeven als een knooppunt en de verbindingen (relaties en interacties) daartussen worden randen genoemd. Door deze verbindingen te analyseren, onderzoekers kunnen veel leren over elke gebruiker - interesses, hobby's, winkelgewoonten, vrienden, enzovoort.
In de biologie, vergelijkbare algoritmen voor het clusteren van grafieken kunnen worden gebruikt om de eiwitten te begrijpen die de meeste functies van het leven uitvoeren. Naar schatting bevat het menselijk lichaam alleen al ongeveer 100, 000 verschillende eiwitsoorten, en bijna alle biologische taken - van spijsvertering tot immuniteit - vinden plaats wanneer deze micro-organismen met elkaar interageren. Een beter begrip van deze netwerken kan onderzoekers helpen de effectiviteit van een medicijn te bepalen of mogelijke behandelingen voor verschillende ziekten te identificeren.
Vandaag, geavanceerde high-throughput-technologieën stellen onderzoekers in staat om honderden miljoenen eiwitten te vangen, genen en andere cellulaire componenten tegelijk en in verschillende omgevingscondities. Clusteringsalgoritmen worden vervolgens toegepast op deze datasets om patronen en relaties te identificeren die kunnen wijzen op structurele en functionele overeenkomsten. Hoewel deze technieken al meer dan tien jaar op grote schaal worden gebruikt, ze kunnen de stortvloed aan biologische gegevens die wordt gegenereerd door de volgende generatie sequencers en microarrays niet bijhouden. In feite, zeer weinig bestaande algoritmen kunnen een biologisch netwerk clusteren dat miljoenen knooppunten (eiwitten) en randen (verbindingen) bevat.
Daarom heeft een team van onderzoekers van het Lawrence Berkeley National Laboratory (Berkeley Lab) en het Joint Genome Institute (JGI) van het Department of Energy (DOE's) een van de meest populaire clusteringbenaderingen in de moderne biologie gekozen - het Markov Clustering (MCL) -algoritme - en aangepast om snel te werken, efficiënt en op schaal op supercomputers met gedistribueerd geheugen. In een testcase, hun krachtige algoritme, HipMCL genaamd, bereikte een voorheen onmogelijke prestatie:het clusteren van een groot biologisch netwerk met ongeveer 70 miljoen knooppunten en 68 miljard randen in een paar uur, met ongeveer 140, 000 processorkernen op de Cori-supercomputer van het National Energy Research Scientific Computing Center (NERSC). Een paper waarin dit werk wordt beschreven, is onlangs gepubliceerd in het tijdschrift Onderzoek naar nucleïnezuren .
"Het echte voordeel van HipMCL is het vermogen om enorme biologische netwerken te clusteren die onmogelijk te clusteren waren met de bestaande MCL-software, waardoor we de nieuwe functionele ruimte in de microbiële gemeenschappen kunnen identificeren en karakteriseren, " zegt Nikos Kyrpides, die aan het hoofd staat van JGI's Microbiome Data Science-inspanningen en het Prokaryote Super Program en is co-auteur van het papier. "Bovendien kunnen we dat doen zonder iets van de gevoeligheid of nauwkeurigheid van de oorspronkelijke methode op te offeren, dat is altijd de grootste uitdaging in dit soort schaalinspanningen."
"Naarmate onze gegevens groeien, het wordt nog belangrijker dat we onze tools verplaatsen naar high-performance computeromgevingen, " voegt hij eraan toe. "Als je me zou vragen hoe groot is de eiwitruimte? De waarheid is, we weten het niet echt, want tot nu toe hadden we niet de rekenhulpmiddelen om al onze genomische gegevens effectief te clusteren en de functionele donkere materie te onderzoeken."
Naast de vooruitgang in de technologie voor gegevensverzameling, onderzoekers kiezen er steeds vaker voor om hun gegevens te delen in gemeenschapsdatabases zoals het Integrated Microbial Genomes &Microbiomes (IMG/M)-systeem, die is ontwikkeld door een decennia-oude samenwerking tussen wetenschappers van JGI en de Computational Research Division (CRD) van Berkeley Lab. Maar door gebruikers in staat te stellen vergelijkende analyses uit te voeren en de functionele mogelijkheden van microbiële gemeenschappen te verkennen op basis van hun metagenomische sequentie, communitytools zoals IMG/M dragen ook bij aan de data-explosie in technologie.
Hoe willekeurige wandelingen leiden tot computerproblemen
Om grip te krijgen op deze datastroom, onderzoekers vertrouwen op clusteranalyse, of clusteren. Dit is in wezen de taak van het groeperen van objecten, zodat items in dezelfde groep (cluster) meer op elkaar lijken dan die in andere clusters. Al meer dan een decennium, computationele biologen hebben de voorkeur gegeven aan MCL voor het clusteren van eiwitten door overeenkomsten en interacties.
"Een van de redenen dat MCL populair is onder computationele biologen, is dat het relatief parametervrij is; gebruikers hoeven niet veel parameters in te stellen om nauwkeurige resultaten te krijgen en het is opmerkelijk stabiel voor kleine wijzigingen in de gegevens. Dit is belangrijk omdat u misschien een overeenkomst tussen gegevenspunten opnieuw moet definiëren of dat u een kleine meetfout in uw gegevens moet corrigeren. u wilt niet dat uw wijzigingen de analyse wijzigen van 10 clusters naar 1, 000 clusters, " zegt Aydin Buluç, een CRD-wetenschapper en een van de co-auteurs van het artikel.
Maar, hij voegt toe, de computationele biologiegemeenschap stuit op een computerknelpunt omdat de tool meestal op een enkel computerknooppunt draait, is rekenkundig duur om uit te voeren en heeft een grote geheugenvoetafdruk - die allemaal de hoeveelheid gegevens beperken die dit algoritme kan clusteren.
Een van de meest rekenkundige en geheugenintensieve stappen in deze analyse is een proces dat random walk wordt genoemd. Deze techniek kwantificeert de sterkte van een verbinding tussen knooppunten, wat handig is voor het classificeren en voorspellen van koppelingen in een netwerk. In het geval van een zoekopdracht op internet, dit kan u helpen een goedkope hotelkamer in San Francisco te vinden voor de voorjaarsvakantie en u zelfs vertellen wat de beste tijd is om deze te boeken. In de biologie, zo'n hulpmiddel kan u helpen bij het identificeren van eiwitten die uw lichaam helpen een griepvirus te bestrijden.
Gegeven een willekeurige grafiek of netwerk, het is moeilijk om de meest efficiënte manier te weten om alle knooppunten en links te bezoeken. Een willekeurige wandeling krijgt een idee van de voetafdruk door de hele grafiek willekeurig te verkennen; het begint bij een knoop en beweegt willekeurig langs een rand naar een naburig knooppunt. Dit proces gaat door totdat alle knooppunten in het grafennetwerk zijn bereikt. Omdat er veel verschillende manieren zijn om tussen knooppunten in een netwerk te reizen, deze stap herhaalt zich vele malen. Algoritmen zoals MCL zullen dit random walk-proces blijven uitvoeren totdat er geen significant verschil meer is tussen de iteraties.
In een willekeurig netwerk kan je hebt misschien een knooppunt dat is verbonden met honderden knooppunten en een ander knooppunt met slechts één verbinding. De willekeurige wandelingen zullen de sterk verbonden knooppunten vastleggen omdat er elke keer dat het proces wordt uitgevoerd een ander pad wordt gedetecteerd. Met deze informatie, het algoritme kan met een zekere mate van zekerheid voorspellen hoe een knooppunt op het netwerk met een ander knooppunt is verbonden. Tussen elke willekeurige wandeling door, het algoritme markeert zijn voorspelling voor elk knooppunt in de grafiek in een kolom van een Markov-matrix - een beetje zoals een grootboek - en aan het einde worden de uiteindelijke clusters onthuld. Het klinkt eenvoudig genoeg, maar voor eiwitnetwerken met miljoenen knooppunten en miljarden randen, dit kan een extreem rekenkundig en geheugenintensief probleem worden. Met HipMCL, Computerwetenschappers van Berkeley Lab gebruikten geavanceerde wiskundige hulpmiddelen om deze beperkingen te overwinnen.
"We hebben met name de MCL-ruggengraat intact gehouden, waardoor HipMCL een massaal parallelle implementatie is van het originele MCL-algoritme, " zegt Ariful Azad, een computerwetenschapper in CRD en hoofdauteur van het artikel.
Hoewel er eerdere pogingen zijn geweest om het MCL-algoritme parallel te laten lopen op een enkele GPU, de tool kon nog steeds alleen relatief kleine netwerken clusteren vanwege geheugenbeperkingen op een GPU, Azad merkt op.
"Met HipMCL herwerken we in wezen de MCL-algoritmen om efficiënt te werken, parallel op duizenden processors, en instellen om te profiteren van het totale geheugen dat beschikbaar is in alle rekenknooppunten, " voegt hij eraan toe. "De ongekende schaalbaarheid van HipMCL komt van het gebruik van ultramoderne algoritmen voor schaarse matrixmanipulatie."
Volgens Buluç, het gelijktijdig uitvoeren van een willekeurige wandeling vanuit veel knooppunten van de grafiek kan het beste worden berekend met behulp van sparse-matrix matrixvermenigvuldiging, wat een van de meest elementaire bewerkingen is in de onlangs uitgebrachte GraphBLAS-standaard. Buluç en Azad ontwikkelden enkele van de meest schaalbare parallelle algoritmen voor GraphBLAS' sparse-matrix matrixvermenigvuldiging en wijzigden een van hun ultramoderne algoritmen voor HipMCL.
"De crux hier was om de juiste balans te vinden tussen parallellisme en geheugenconsumptie. HipMCL extraheert dynamisch zoveel mogelijk parallellisme gezien het beschikbare geheugen dat eraan is toegewezen, " zegt Buluç.
HipMCL:clustering op schaal
Naast de wiskundige innovaties, een ander voordeel van HipMCL is de mogelijkheid om naadloos te werken op elk systeem, inclusief laptops, werkstations en grote supercomputers. De onderzoekers bereikten dit door hun tools in C++ te ontwikkelen en standaard MPI- en OpenMP-bibliotheken te gebruiken.
"We hebben HipMCL uitgebreid getest op Intel Haswell, Ivy Bridge en Knights Landing-processors bij NERSC, met behulp van een maximaal 2, 000 nodes en een half miljoen threads op alle processors, en in al deze runs heeft HipMCL met succes geclusterde netwerken bestaande uit duizenden tot miljarden randen, " zegt Buluç. "We zien dat er geen barrière is in het aantal processors dat het kan gebruiken om te draaien en ontdekken dat het netwerken 1 kan clusteren, 000 keer sneller dan het originele MCL-algoritme."
"HipMCL zal echt transformerend zijn voor de computationele biologie van big data, net zoals de IMG- en IMG/M-systemen zijn geweest voor microbioomgenomica, " zegt Kyrpides. "Deze prestatie is een bewijs van de voordelen van interdisciplinaire samenwerking bij Berkeley Lab. Als biologen begrijpen we de wetenschap, maar het is van zo onschatbare waarde geweest om samen te kunnen werken met computerwetenschappers die ons kunnen helpen onze beperkingen aan te pakken en ons vooruit te stuwen."
Hun volgende stap is om door te gaan met het herwerken van HipMCL en andere computationele biologietools voor toekomstige exascale-systemen, die in staat zal zijn om triljoen berekeningen per seconde te berekenen. Dit zal essentieel zijn aangezien genomics-gegevens in een verbijsterend tempo blijven groeien - ongeveer elke vijf tot zes maanden verdubbelen. Dit zal worden gedaan als onderdeel van het Exagraph co-designcentrum van het DOE Exascale Computing Project.
 Onderzoekers ontdekken nieuwe structuur voor veelbelovende materiaalklasse
Onderzoekers ontdekken nieuwe structuur voor veelbelovende materiaalklasse- Antikankermechanisme onthuld in gistexperimenten
- Vrijstaande fotoverknoopte eiwitpolymeerhydrogels voor langdurige afgifte van geneesmiddelen
- Forensische onderzoekers vinden een nauwkeurigere manier om de leeftijd van overledenen te schatten
- Wetenschappers visualiseren de structuur van een sleutelenzym dat triglyceriden maakt
- Onomkeerbaar omslagpunt van de opwarming mogelijk geactiveerd:hoofd van de Arctische missie
- NASA ziet orkaan Michael landinwaarts trekken
- El Nino is snel sterker en vreemder geworden, volgens koraalrecords
- Hoe reproduceren platwormen en rondwormen?
- Antropologen doorzoeken NASA-gegevens op patronen van migratie en landgebruik in de Himalaya
Hoofdlijnen
- Difference Between Mutation & Genetic Drift
- Onderzoek bevestigt het:we worden echt dommer
- Geen zoetekauw meer? Wetenschap schakelt het verlangen naar suiker bij muizen uit
- Boek beschrijft alle 451 families van bloeiende planten, varens, lycopoden en naaktzadigen
- Earth Microbiome Project:het microbioom van... alles in kaart brengen
- Hoe huilen werkt
- De functie van macromoleculen
- Een natuurlijke schimmelstam kan olielekkages opruimen en de oliezanden van Albertas weer tot leven brengen
- Onderzoek toont belang aan van dieren in het wild bij het bestrijden van teken
- 10 redenen waarom u zich zorgen zou moeten maken over gezichtsherkenningstechnologie

- EU wil internetbedrijven dwingen terreurinhoud te verwijderen

- grote vervoerders, staat AG's zullen werken om robocalls te bestrijden

- CrocSpotter-app gebruikt AI voor detectie

- Musk overhandigt in China gemaakte Tesla's aan vroege kopers in Shanghai


 Overblijfselen gegraven uit Japans massagraf suggereren epidemie in 1800
Overblijfselen gegraven uit Japans massagraf suggereren epidemie in 1800- Natuurlijke klimaatpatronen creëren hotspots van snelle zeespiegelstijging
- Kleurstofgevoelige zonnecel absorbeert een breed scala aan zichtbare en infrarode golflengten
- Hoe sommige landen het door het coronavirus getroffen toerisme in Afrika nieuw leven inblazen
- De huidige opkomst van extreemrechts begrijpen met behulp van Marx en Lacan
- Aan een zijden draadje hangen:beeldvorming en sondeerketens van enkele atomen
- Hoe werkt koolstofvastlegging?
- Waarom vormt condensatie zich op een drinkglas?
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
