Wetenschap
Een diepe variatie-autoencoder voor proteomics-massaspectrometriegegevensanalyse
Het team van Jianwei Shuai en het team van Jiahuai Han aan de Universiteit van Xiamen hebben een diepgaande, op autoencoder gebaseerde, data-onafhankelijke acquisitiedata-analysesoftware voor eiwitmassaspectrometrie ontwikkeld, die de analyse van relevante peptiden en eiwitten uit complexe eiwitmassaspectrometriegegevens realiseert, en de superioriteit en demonstreert veelzijdigheid van de methode op verschillende instrumenten en soortenmonsters. Het onderzoek is gepubliceerd in Research als "Beste-DIA XMBD :diepe autoencoder voor data-onafhankelijke acquisitieproteomics".
Eiwitten spelen een cruciale rol als uitvoerders van cellulaire levensactiviteiten en sturen een groot aantal cruciale biologische processen aan. Bijgevolg heeft het gebied van proteomics brede aandacht gekregen. Proteomics omvat de uitgebreide studie van eiwiteigenschappen, inclusief post-translationele modificaties, eiwitexpressieniveaus, eiwit-eiwitinteracties en meer. Het algemene doel is om een holistisch inzicht te krijgen in de pathogenese van ziekten, het cellulaire metabolisme en andere vitale processen op eiwitniveau.
Van de belangrijkste analytische technieken in proteomisch onderzoek springt eiwitmassaspectrometrie eruit als de meest kritische. In de loop van de tijd is de massaspectrometrietechnologie geëvolueerd om onderzoekers te voorzien van betrouwbare en dynamische hulpmiddelen voor proteomics-analyse.
Twee belangrijke benaderingen van eiwitmassaspectrometrie zijn data-afhankelijke acquisitie (DDA) en data-onafhankelijke acquisitie (DIA). Bij DDA worden alle peptideprecursor-ionspectra (MS1) in volledige scanmodus verkregen, gevolgd door selectie van de meest N-intensieve peptide-ionen voor fragmentatie om fragmentionspectra (MS2) te verkrijgen.
Ondanks het nut ervan wordt DDA geconfronteerd met uitdagingen die verband houden met experimentele reproduceerbaarheid en detectie van peptiden met een lage overvloed, als gevolg van de willekeur van peptidefragmentatie en de preferentiële selectie van peptiden met hoge intensiteit.
Om deze beperkingen te overwinnen is de DIA-acquisitiemethode geïntroduceerd. Deze techniek verdeelt het massa-ladingsverhoudingsbereik van ouderionspectra in meerdere vensters en fragmenteert achtereenvolgens alle peptiden binnen elk venster om dochterionspectra te verkrijgen. Een veelgebruikte DIA-methode is Sequential Window Acquisition of all Theoretical fragmentionen (SWATH).
Hoewel DIA-acquisitiegegevens uitgebreidere proteomische informatie behouden, vormen de grote gegevensgrootte, hoge dimensionaliteit en complexe spectrale signalen uitdagingen voor de analyse ervan. Als gevolg hiervan is DIA-datamining een belangrijk aandachtspunt geworden in de proteomics-gemeenschap.
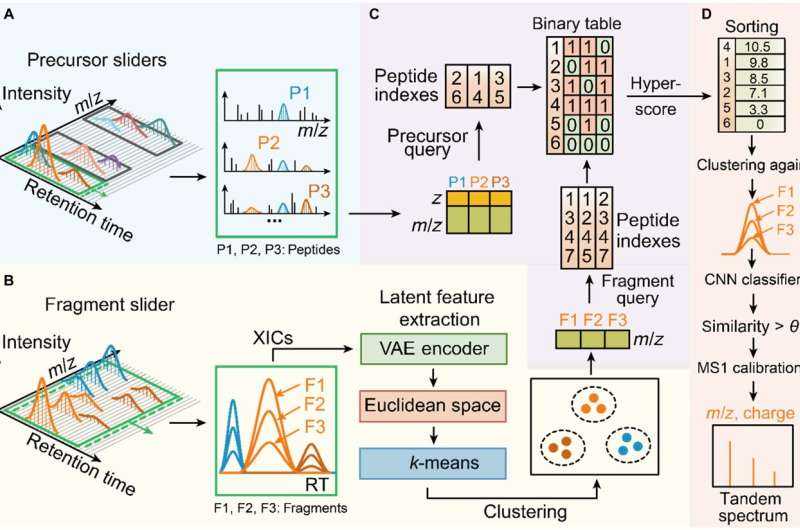
Het team van Jianwei Shuai en het team van Jiahuai Han hebben samengewerkt om Dear-DIA te ontwikkelen, een op deep learning gebaseerde data-onafhankelijke software voor acquisitiedata-analyse, die de identificatie realiseert van fragmentionen die overeenkomen met verschillende peptiden uit complexe DIA-acquisitiespectra en de generalisatie naar complexe monsters demonstreert. van verschillende soorten.
Dear-DIA verdeelt de spectra eerst in een schuifregelaar met vaste breedte en een vaste breedte langs de retentietijd (RT) richting, en elke schuifregelaar bevat een reeks voorloperspectra MS1 en fragmentspectra MS2 als de minimale verwerkingseenheid. Vervolgens werd een algoritme voor het vinden van pieken gebruikt om de achtergrondionen met een laag signaal-ruisniveau te verwijderen en de kandidaat-voorloperionen en kandidaat-fragmentionen vast te houden.
Vervolgens gebruikt Dear-DIA een variatie-autoencoder om de piekkenmerken van fragmentionen te extraheren en brengt de kenmerken in de Euclidische ruimte in kaart, en clustert vervolgens de kenmerken, met verschillende klassen van fragmenten die overeenkomen met verschillende peptiden, waardoor het deconvolutieproces van het spectrogram wordt gerealiseerd. P>
Dear-DIA bevat een indexeringsalgoritme genaamd PIndex, dat de voorlopers vergelijkt met de clusterresultaten van fragmenten en de beste koppelingsresultaten selecteert door te scoren. Dear-DIA gebruikt een convolutioneel neuraal netwerk om de gelijkenis van de piekvorm van fragmenten in dezelfde klasse opnieuw te berekenen om interfererende ionen en clusterresultaten met een lage gelijkenis te elimineren.
De auteurs testten eerst de prestaties van Dear-DIA op een SGS Human-dataset met 422 synthetische peptiden van stabiele isotoop-gelabelde standaarden verdeeld in 10 verdunningsgradiënten (van 1-voudige tot 512-voudige verdunning), en DIA-gegevens werden verkregen op een AB SCIEX TTOF5600 massaspectrometer die de SWATH-techniek gebruikt om DIA-gegevens te verkrijgen.
Uit de analyseresultaten bleek dat Dear-DIA meer synthetische peptiden aantrof in alle verdunde oplossingen vergeleken met de twee veelgebruikte analysemethoden, Spectronaut 14 en DIA-Umpire. De auteurs vergeleken ook het aantal peptiden en eiwitten dat werd gevonden met de verschillende analytische methoden voor de datasets van SGS Human en L929 Mouse. Uit de resultaten bleek dat Dear-DIA meer peptiden en eiwitten kon vinden vergeleken met Spectronaut 14 en DIA-Umpire, wat neerkomt op meer dan 85% van hun resultaten.
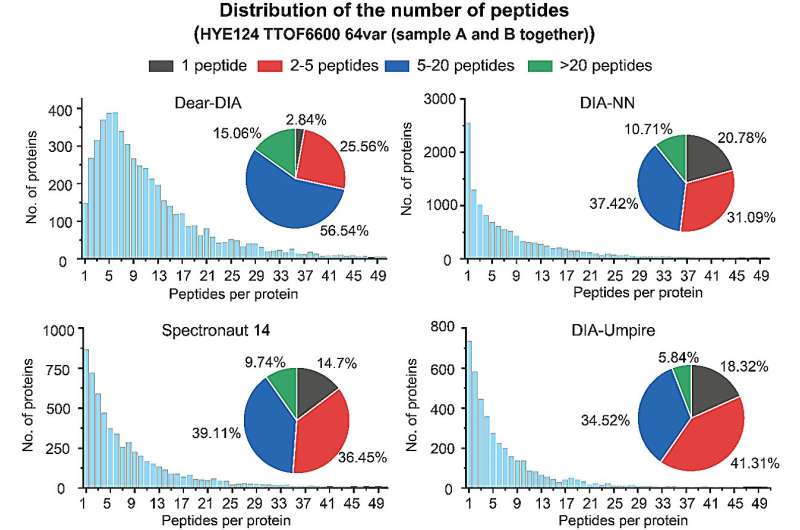
Het vertrouwen van de resultaten van proteomics-analyse kan ook worden aangetoond door het aantal peptiden dat voor elk eiwit is geïdentificeerd. Eiwitten met twee of meer geïdentificeerde peptiden worden over het algemeen als geloofwaardiger identificaties beschouwd. De auteurs vergeleken het door Dear-DIA gerapporteerde aantal eiwitten versus peptiden met bestaande software op een dataset met gemengde soorten (HYE124 TTOF6600 64var dataset).
De dataset bevat eiwitten van drie soorten, mens, gist en E. coli, en de gegevens zijn verkregen op een AB SCIEX TTOF6600 massaspectrometer met behulp van de SWATH-methode, met ouderionspectra die 64 variabele vensters bevatten. Uit de analyseresultaten bleek dat 97,16% van de door Dear-DIA gevonden eiwitten konden overeenkomen met 2 of meer peptiden, wat veel hoger is dan DIA-NN, Spectronaut 14 en DIA-Umpire.
Data-onafhankelijke acquisitietechnieken voor proteomics zijn op grote schaal toegepast en gerelateerde analyse-algoritmen zijn een hotspot voor onderzoek geworden. Eiwitontdekking uit massaspectrometriegegevens is een interessante en uitdagende taak. In dit artikel ontwikkelde het team Dear-DIA, een analysesoftware gebaseerd op deep learning, die wordt gebruikt om een verscheidenheid aan zeer complexe DIA-acquisitiegegevens te verwerken en meer peptiden en eiwitten kan ontdekken, naast het reproduceren van de meeste resultaten van Spectronaut en DIA-scheidsrechter.
Hoewel de trainingsdataset afkomstig is van E. coli, demonstreert de uitstekende prestatie van Dear-DIA op de dataset met gemengde soorten bovendien het sterke generalisatievermogen ervan om complexe proteomicsgegevens te analyseren. Deep learning, als veelgebruikt hulpmiddel voor big data-analyse, heeft uitstekende datamining-mogelijkheden aangetoond om diepgaande intrinsieke associaties in big data te ontdekken.
Het gebruik van deep learning om proteomics massaspectrometriegegevens te analyseren heeft een groot potentieel en zal de studie van fundamentele kwesties zoals eiwitsignaleringsnetwerken verder bevorderen.
Meer informatie: Qingzu He et al., Dear-DIA XMBD :Deep Autoencoder maakt deconvolutie van data-onafhankelijke acquisitie-proteomics mogelijk, Onderzoek (2023). DOI:10.34133/onderzoek.0179
Journaalinformatie: Onderzoek
Geleverd door Onderzoek
 gevaren in kaart brengen, geschiedenis en de toekomst van Rust Belt-steden
gevaren in kaart brengen, geschiedenis en de toekomst van Rust Belt-steden- Giftige afvoer van de Tijuana-rivier valt Imperial Beach binnen
- Amerikaanse staat geeft belangrijke goedkeuring voor Keystone-pijplijn
- Wat telt als natuur? Het hangt er vanaf
- Ruimtelijke variatie in carbonaatkoolstofisotopen tijdens Cambrian SPICE-evenement in het oosten van Noord-China
Hoofdlijnen
- Rangschikken met behulp van een vierkantswortelcurve
- Ribosomen: definitie, functie en structuur (eukaryoten en prokaryoten)
- Australisch onderzoek bewijst dat mensen het meest angstaanjagende roofdier van de planeet zijn
- Onderzoek naar zeevogels aan de westkust laat zien hoe ze op een dag de lucht zouden kunnen delen met windturbines
- Onderzoekers stellen het gebruik van elektrische stroomuitval voor om de impact van kunstlicht op dieren in het wild te bepalen
- Wat is het voordeel van het gebruik van vlekken om naar cellen te kijken?
- Wetenschappers gebruiken bladgloed om het veranderende klimaat te begrijpen
- Bio-engineered enzym creëert in één stap natuurlijke vanilline uit planten
- Fossil vangt zeesterren die zichzelf in tweeën splitsen – wat aantoont dat dit al 155 miljoen jaar gebeurt
- Natuurkundigen gebruiken wiskundige algoritmen om experimentele 3D-structuren van chromosomen te onderzoeken
- Bananenschillen maken suikerkoekjes beter voor je

- Chemici hebben de oorsprong van de groene fluorescentie uitgelegd

- Katalysator zet plastic afval om in waardevolle ingrediënten bij lage temperatuur

- Vormverschuivend materiaal kan morphen, keert zichzelf om met behulp van warmte, licht


 Kennis van vreemde talen gaat een leven lang mee, blijkt uit nieuw onderzoek
Kennis van vreemde talen gaat een leven lang mee, blijkt uit nieuw onderzoek- Gevaar niet voorbij:gaslekken, schimmel weefgetouw voor Harvey evacués
- Kleine gouden roosters die geheimen opleveren
- 3D-print een stukje Mars voor de feestdagen
- Demografie van deportatie:niet-burgers doen het beter in gemeenschappen die 20-40 procent Spaans zijn
- Canyon in Arizona, beroemd om watervallen die na overstromingen weer opengaan
- Door gletsjers gevoede rivieren kunnen atmosferische kooldioxide verbruiken
- Alle basismeeteenheden zijn nu gekoppeld aan gedefinieerde constanten in plaats van fysieke objecten
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
