Wetenschap
Machine learning framework ID's doelen voor het verbeteren van katalysatoren
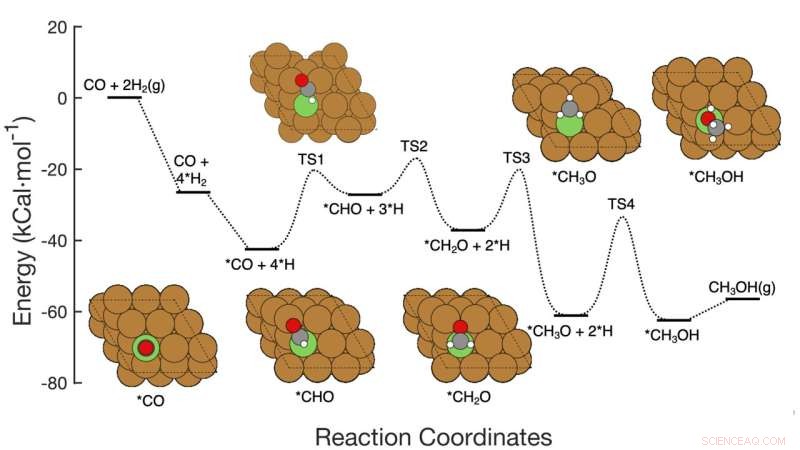
Deze afbeelding toont de zevenstapsreactieroute van CO-hydrogenering tot methanol over op koper gebaseerde katalysatoren, inclusief de reactanten bij elke stap, schematische atomaire rangschikkingen van de tussenproducten en de energieactiveringsbarrières die nodig zijn om van stap naar stap te gaan. Het Brookhaven Lab-team demonstreerde een machine learning-raamwerk dat met succes identificeerde welke stappen/combinaties van stappen moesten worden aangepast om de methanolproductie te verbeteren. Hun werk zou kunnen helpen bij het ontwerpen van nieuwe katalysatoren om dat doel te bereiken en het raamwerk kan worden toegepast om andere reacties te optimaliseren. Krediet:Brookhaven National Laboratory
Chemici van het Brookhaven National Laboratory van het Amerikaanse ministerie van Energie hebben een nieuw machinaal leren (ML) -raamwerk ontwikkeld dat kan bepalen welke stappen van een meerstaps chemische conversie moeten worden aangepast om de productiviteit te verbeteren. De aanpak zou kunnen helpen bij het ontwerpen van katalysatoren - chemische 'dealmakers' die reacties versnellen.
Het team ontwikkelde de methode om de omzetting van koolmonoxide (CO) in methanol te analyseren met behulp van een op koper gebaseerde katalysator. De reactie bestaat uit zeven vrij eenvoudige elementaire stappen.
"Ons doel was om te identificeren welke elementaire stap in het reactienetwerk of welke subset van stappen de katalytische activiteit regelt", zegt Wenjie Liao, de eerste auteur van een paper waarin de methode wordt beschreven die zojuist is gepubliceerd in het tijdschrift Catalysis Science &Technology . Liao is een afgestudeerde student aan de Stony Brook University die heeft gewerkt met wetenschappers in de Catalysis Reactivity and Structure (CRS) -groep in de Chemistry Division van Brookhaven Lab.
Ping Liu, de CRS-chemicus die het werk leidde, zei:"We gebruikten deze reactie als voorbeeld van onze ML-raamwerkmethode, maar je kunt elke reactie in het algemeen in dit raamwerk plaatsen."
Activeringsenergie targeten
Stel je een chemische reactie in meerdere stappen voor als een achtbaan met heuvels van verschillende hoogtes. De hoogte van elke heuvel vertegenwoordigt de energie die nodig is om van de ene trede naar de volgende te komen. Katalysatoren verlagen deze "activeringsbarrières" door het voor reactanten gemakkelijker te maken om samen te komen of door ze dat te laten doen bij lagere temperaturen of drukken. Om de algehele reactie te versnellen, moet een katalysator zich richten op de stap of stappen die de grootste impact hebben.
Traditioneel zouden wetenschappers die een dergelijke reactie willen verbeteren, berekenen hoe elke activeringsbarrière één tegelijk . moet worden gewijzigd kan de totale productiesnelheid beïnvloeden. Dit type analyse zou kunnen identificeren welke stap "snelheidsbeperkend" was en welke stappen de selectiviteit van de reactie bepalen, dat wil zeggen of de reactanten naar het gewenste product gaan of een alternatieve route naar een ongewenst bijproduct volgen.

Brookhaven Lab-chemicus Ping Liu en Wenjie Liao, een afgestudeerde student aan de Stony Brook University, ontwikkelden een raamwerk voor machinaal leren om te identificeren welke chemische reactiestappen zouden kunnen worden gericht om de reactieproductiviteit te verbeteren. Krediet:Brookhaven National Laboratory
Maar volgens Liu:"Deze schattingen zijn uiteindelijk erg ruw met veel fouten voor sommige groepen katalysatoren. Dat heeft echt pijn gedaan voor het ontwerp en de screening van de katalysator, en dat is wat we proberen te doen," zei ze.
Het nieuwe raamwerk voor machine learning is ontworpen om deze schattingen te verbeteren, zodat wetenschappers beter kunnen voorspellen hoe katalysatoren de reactiemechanismen en de chemische output zullen beïnvloeden.
"In plaats van één barrière tegelijk te verplaatsen, verplaatsen we nu alle barrières tegelijk. En we gebruiken machine learning om die dataset te interpreteren", zegt Liao.
Deze aanpak geeft volgens het team veel betrouwbaardere resultaten, ook over hoe stappen in een reactie samenwerken.
"Onder reactieomstandigheden zijn deze stappen niet geïsoleerd of van elkaar gescheiden; ze zijn allemaal met elkaar verbonden", zei Liu. "Als je maar één stap tegelijk doet, mis je veel informatie - de interacties tussen de elementaire stappen. Dat is wat in deze ontwikkeling is vastgelegd", zei ze.
Het model bouwen
De wetenschappers begonnen met het bouwen van een dataset om hun machine learning-model te trainen. De dataset was gebaseerd op "density functional theory" (DFT) berekeningen van de activeringsenergie die nodig is om de ene rangschikking van atomen naar de volgende te transformeren door de zeven stappen van de reactie. Vervolgens voerden de wetenschappers computergebaseerde simulaties uit om te onderzoeken wat er zou gebeuren als ze alle zeven activeringsbarrières tegelijkertijd zouden veranderen - sommige gaan omhoog, sommige gaan omlaag, sommige afzonderlijk en sommige in paren.
"De reeks gegevens die we hebben opgenomen, was gebaseerd op eerdere ervaringen met deze reacties en dit katalytische systeem, binnen het interessante variatiebereik dat u waarschijnlijk betere prestaties zal geven", zei Liu.
Door variaties te simuleren in 28 "descriptoren" - inclusief de activeringsenergieën voor de zeven stappen plus paren stappen die er twee tegelijk veranderen - produceerde het team een uitgebreide dataset van 500 datapunten. Deze dataset voorspelde hoe al die individuele tweaks en paren tweaks de methanolproductie zouden beïnvloeden. Het model scoorde vervolgens de 28 descriptoren op basis van hun belang bij het stimuleren van de methanolproductie.
"Ons model heeft 'geleerd' van de gegevens en identificeerde zes belangrijke descriptoren waarvan het voorspelt dat ze de meeste impact zullen hebben op de productie," zei Liao.
Nadat de belangrijke descriptoren waren geïdentificeerd, hebben de wetenschappers het ML-model opnieuw getraind met alleen die zes "actieve" descriptoren. Dit verbeterde ML-model was in staat om katalytische activiteit te voorspellen, puur gebaseerd op DFT-berekeningen voor die zes parameters.
"In plaats van dat je de hele 28 descriptoren moet berekenen, kun je nu met alleen de zes descriptoren rekenen en de methanolconversiepercentages krijgen waarin je geïnteresseerd bent," zei Liu.
Het team zegt dat ze het model ook kunnen gebruiken om katalysatoren te screenen. Als ze een katalysator kunnen ontwerpen die de waarde van de zes actieve descriptoren verbetert, voorspelt het model een maximale methanolproductie.
Mechanismen begrijpen
Toen het team de voorspellingen van hun model vergeleek met de experimentele prestaties van hun katalysator - en de prestaties van legeringen van verschillende metalen met koper - kwamen de voorspellingen overeen met de experimentele bevindingen. Vergelijkingen van de ML-benadering met de vorige methode die werd gebruikt om de prestaties van legeringen te voorspellen, toonden aan dat de ML-methode veel beter was.
De gegevens onthulden ook veel details over hoe veranderingen in energiebarrières het reactiemechanisme zouden kunnen beïnvloeden. Van bijzonder belang - en belangrijk - was hoe verschillende stappen van de reactie samenwerken. De gegevens toonden bijvoorbeeld aan dat in sommige gevallen het verlagen van de energiebarrière in de snelheidsbeperkende stap alleen op zichzelf de methanolproductie niet zou verbeteren. Maar het aanpassen van de energiebarrière van een stap eerder in het reactienetwerk, terwijl de activeringsenergie van de snelheidsbeperkende stap binnen een ideaal bereik blijft, zou de methanoloutput verhogen.
"Onze methode geeft ons gedetailleerde informatie die we mogelijk kunnen gebruiken om een katalysator te ontwerpen die de interactie tussen deze twee stappen goed coördineert," zei Liu.
Maar Liu is het meest enthousiast over het potentieel om dergelijke datagestuurde ML-frameworks toe te passen op meer gecompliceerde reacties.
"We hebben de methanolreactie gebruikt om onze methode te demonstreren. Maar de manier waarop het de database genereert en hoe we het ML-model trainen en hoe we de rol van de functie van elke descriptor interpoleren om het totale gewicht in termen van hun belang te bepalen - dat kan zijn gemakkelijk toegepast op andere reacties," zei ze. + Verder verkennen
Ontdekking van een nieuwe katalysator voor zeer actieve en selectieve hydrogenering van kooldioxide tot methanol
 Een 20% -suikeroplossing maken
Een 20% -suikeroplossing maken- Wat zijn de kleuren van een vuur en hoe heet zijn ze?
- Maak een lijst van enkele factoren die de diffusiesnelheid zouden verhogen
- Voedingswetenschappers maken gezonde probiotische drank van sojapulp
- Mysterie van hoe zwarte weduwe-spinnen ijzersterke zijden webben creëren, verder ontrafeld
- Hoe beschermen haaien zichzelf?
- Duizenden vast op door vuur omringde stranden in Australië
- Langetermijnstudie onthult voordelen voor de volksgezondheid door vermindering van luchtvervuiling
- Microbiële gemeenschappen hebben een seizoensverandering doorgemaakt
- De winning van fossiele brandstoffen beperken om de opwarming van de aarde onder de 1,5°C-doelstelling te houden
Hoofdlijnen
- Het gebruik van pinguïns om de gezondheid van de oceaan te monitoren is mogelijk niet effectief
- Verschillen tussen "Fysiek" en "Fysiologisch"
- Hoe een bloeiende legale marihuana-industrie de luchtkwaliteit kan schaden
- Herstel van veengebieden:overstromingen zijn niet de ideale oplossing
- Hoe reageert het excretiesysteem op fysieke activiteit?
- Wetenschappers ontdekken pad naar verbeterde gerstkwaliteit
- Hoe creëren de hersenen een ononderbroken kijk op de wereld?
- Your Body On: A Horror Movie
- Toenemend bewijs dat beren geen carnivoren zijn
- Isopropanol Alcohol Vs. Isopropylalcohol

- Onderzoekers ontwikkelen met goud gecomplexeerde ferrocenylfosfines als krachtige antimalariamiddelen
- Snelle detectie van toxische verbindingen
- Nieuwe MD-simulatie werpt licht op het mysterie van de structuur van gehydrateerde elektronen

- Van de bronstijd tot voedselblikken, hier is hoe tin de mensheid heeft veranderd


 Speekseleiwitten kunnen verklaren waarom sommige mensen te veel zout gebruiken
Speekseleiwitten kunnen verklaren waarom sommige mensen te veel zout gebruiken- Online risico's zijn routine voor tieners, meest stuiteren terug
- Cilia: definitie, types en functie
- Wat de wereld kan leren van de verwoestende overstromingen in Pakistan
- Licht kan niet-magnetische metalen magnetiseren, natuurkundigen voorstellen
- Na het in kaart brengen van miljoenen sterrenstelsels, donkere energie-enquête voltooit gegevensverzameling
- CT-scans van Egyptische mummie onthullen nieuwe details over de dood van een cruciale farao
- Wat zijn de juiste omstandigheden voor de autoclaaf?
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
