Wetenschap
Een model dat is getraind om spectroscopische profielen te voorspellen, helpt de structuur van materialen te ontcijferen
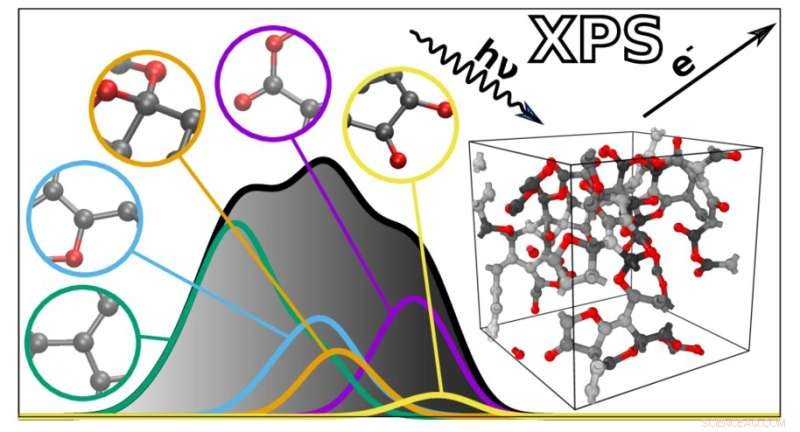
Het nieuwe algoritme voorspelt de XPS-spectra van complexe materialen op basis van individuele atomaire bijdragen. Krediet:Miguel Caro / Aalto University
Op koolstof gebaseerde materialen hebben een enorm potentieel voor het bouwen van een duurzame toekomst, maar materiaalwetenschappers hebben hulpmiddelen nodig om hun atomaire structuur, die hun functionele eigenschappen bepaalt, goed te analyseren. Röntgenfoto-elektronspectroscopie (XPS) is een van de instrumenten die hiervoor worden gebruikt, maar XPS-resultaten kunnen moeilijk te interpreteren zijn. Nu hebben onderzoekers van Aalto een machine-learning tool ontwikkeld om XPS-analyses te verbeteren, die ze vrij beschikbaar hebben gesteld als de XPS Prediction Server.
XPS-spectra zijn grafieken met een verzameling pieken die de bindingsenergie weerspiegelen van de elektronen diep in de atomen waaruit een materiaal bestaat. Omdat de bindingsenergieën afhankelijk zijn van de atomaire omgeving, kunnen ze worden gebruikt om af te leiden hoe de atomen zijn verbonden in een bepaald materiaal of molecuul. Dit maakt XPS-spectra echter ook moeilijk te interpreteren, omdat veel factoren van invloed zijn op bindingsenergieën. De bindingsenergieën van verschillende atomaire kenmerken kunnen elkaar ook overlappen, wat de analyse verder compliceert.
Om hierbij te helpen, ontwikkelde een team onder leiding van Miguel Caro een rekenmethode die het bindingsenergiespectrum van een materiaal kan voorspellen op basis van een computergegenereerd structureel model. Dit vereenvoudigt de interpretatie van XPS-gegevens door het mogelijk te maken om de experimenteel waargenomen bindingsenergieën te vergelijken met de computationele voorspellingen.
Het idee zelf is niet nieuw, maar het probleem was de rekenkundige moeilijkheid om het XPS-spectrum van een materiaal nauwkeurig te berekenen. Het team van Caro heeft dit opgelost met behulp van machine learning. De truc was om een goedkoop computeralgoritme te trainen om de uitkomst van een rekenkundig dure referentiemethode te voorspellen op basis van een efficiënte combinatie van rekenkundig goedkope en dure kwantummechanische gegevens.
De rekenkundig goedkopere methode, DFT, komt niet erg nauwkeurig overeen met de experimentele resultaten. De nauwkeuriger methode, GW, duurt te lang om te berekenen wanneer een molecuul veel atomen heeft. "We hebben besloten om een basismodel te construeren dat overvloedige DFT-gegevens gebruikt en dit vervolgens te verfijnen met schaarse en kostbare GW-gegevens. En het werkte", zegt Caro.
Het resulterende algoritme kan het spectrum voorspellen van elk ongeordend materiaal gemaakt van koolstof, waterstof en zuurstof. "De voorspelde spectra liggen opmerkelijk dicht bij die welke experimenteel zijn verkregen. Dit opent de deur naar een betere integratie tussen experimentele en computationele karakterisering van materialen", zegt Caro. Vervolgens is het team van plan hun techniek uit te breiden met een breder scala aan materialen en andere soorten spectroscopie.
Het open access-artikel is gepubliceerd in Chemistry of Materials . + Verder verkennen
Diamantachtige koolstof wordt anders gevormd dan werd aangenomen - machine learning maakt de ontwikkeling van een nieuw model mogelijk
 Hoe bacteriën hypothiocyaniet onschadelijk maken, een antimicrobieel wapen van het aangeboren immuunsysteem
Hoe bacteriën hypothiocyaniet onschadelijk maken, een antimicrobieel wapen van het aangeboren immuunsysteem- Uw medische implantaat of voedselverpakking kan ooit gemaakt worden van CBD
- Nieuwe draai aan CRISPR-technologie
- Onderzoekers kwantificeren thermodynamisch samenspel tijdens co-aggregatie van eiwitten
- Vol hete lucht en er trots op:Gasopslag verbeteren met MOF's
- Mosselen en kokkels, schildwachten van de milieutoestand van de Nicaraguaanse kusten
- Vergelijking van mondiale klimatologieën bevestigt opwarming van de mondiale oceaan
- Welke soorten dieren worden aangetroffen in zoetwaterecosystemen?
- Vraag en antwoord:La Ninas terug en het is niet goed voor delen van het droge westen
- Stormslagers Bermuda, dwingt annulering van vluchten af
Hoofdlijnen
- De oorsprong van genen voor het maken van bloemen
- Maatregelen voor welzijn en duurzaamheid heroverwegen van lokale naar mondiale schaal
- Hoe BRCA-genen werken
- Immuunfunctie geremodelleerd door mitochondriale vorm
- Wat is cyberchondrie?
- Soorten instrumenten die worden gebruikt voor het meten van lichaamstemperaturen
- Paarse plant is in de verdediging
- Idaho een stap dichter bij de grootste onderzoekszuivelfabriek in de VS
- Medaka-vissen gebruiken gezichten om verschillende individuen te identificeren
- Niezen cam onthult de beste stoffencombinaties voor stoffen maskers
- Synthetische bedrukte polymeren herkend door DNA
- De waardevolle bijdrage van stress aan de thermische stabiliteit van polykristallijne legeringen met nanokorrels

- Nieuwe methode voor microbiële energieproductie ontdekt

- Nieuw inzicht in celmembranen kan het testen en ontwerpen van geneesmiddelen verbeteren

 Nutty Putty Cave voor en na de tragedie van 2009
Nutty Putty Cave voor en na de tragedie van 2009- Waren op weg naar de maan en misschien naar Mars. Dus van wie zijn ze?
- Het mengen van silicium met andere materialen verbetert de diversiteit van elektronische apparaten op nanoschaal
- Spontane vorming van holle structuren op nanoschaal zou de batterijopslag kunnen vergroten
- Amazon blijft groeien, net als zijn cache met gegevens over jou
- Nieuwe studie vindt dat langer thuis blijven de sleutel was tot technologische verandering in de steentijd 60, 000 jaar geleden
- Microsoft-patch wacht op zero-day-kwetsbaarheid
- Kan kelp de verzuring van de oceaan helpen verlichten?
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
