Wetenschap
Machine learning-benadering kan helpen bij het ontwerpen van industriële processen voor de productie van geneesmiddelen

Een nieuw computersysteem voorspelt de producten van chemische reacties. “De visie is dat je naar een systeem kunt lopen en zeggen:‘Ik wil dit molecuul maken.’ De software vertelt je vanaf welke route je het moet maken, en de machine zal het maken, ’, zegt professor Klays Jensen. Krediet:MIT Nieuws
Wanneer organische chemici een bruikbare chemische verbinding identificeren - een nieuw medicijn, het is bijvoorbeeld aan chemische ingenieurs om te bepalen hoe ze in massa kunnen worden geproduceerd.
Er kunnen 100 verschillende reactiesequenties zijn die hetzelfde eindproduct opleveren. Maar sommige gebruiken goedkopere reagentia en lagere temperaturen dan andere, en misschien wel het belangrijkste, sommige zijn veel gemakkelijker om continu te draaien, met technici die af en toe reagentia bijvullen in verschillende reactiekamers.
historisch, het bepalen van de meest efficiënte en kosteneffectieve manier om een bepaald molecuul te produceren is evenzeer kunst als wetenschap geweest. Maar MIT-onderzoekers proberen dit proces op een meer veilige empirische basis te zetten, met een computersysteem dat getraind is op duizenden voorbeelden van experimentele reacties en dat leert voorspellen wat de belangrijkste producten van een reactie zullen zijn.
Het werk van de onderzoekers verschijnt in het tijdschrift van de American Chemical Society ACS Centrale Wetenschap . Zoals alle machine learning-systemen, die van hen presenteert de resultaten in termen van waarschijnlijkheden. Bij testen, het systeem was in staat om 72 procent van de tijd het belangrijkste product van een reactie te voorspellen; 87 procent van de tijd, het rangschikte het belangrijkste product onder de drie meest waarschijnlijke resultaten.
"Er is duidelijk veel begrepen over reacties vandaag, " zegt Klavs Jensen, de Warren K. Lewis Professor of Chemical Engineering aan het MIT en een van de vier senior auteurs op het papier, "maar het is een sterk geëvolueerd, verworven vaardigheid om naar een molecuul te kijken en te beslissen hoe je het gaat synthetiseren uit uitgangsmaterialen."
Met het nieuwe werk Jensen zegt, "de visie is dat je naar een systeem kunt lopen en zeggen:'Ik wil dit molecuul maken.' De software vertelt je vanaf welke route je hem moet maken, en de machine zal het maken."
Met een kans van 72 procent om het belangrijkste product van een reactie te identificeren, het systeem is nog niet klaar om het type volledig geautomatiseerde chemische synthese te verankeren dat Jensen voor ogen heeft. Maar het zou chemische ingenieurs kunnen helpen om sneller de beste volgorde van reacties te vinden - en mogelijk sequenties suggereren die ze anders misschien niet zouden hebben onderzocht.
Jensen wordt op het papier vergezeld door eerste auteur Connor Coley, een afgestudeerde student chemische technologie; Willem Groen, de Hoyt C. Hottel hoogleraar chemische technologie, WHO, met Jensen, co-adviseert Coley; Regina Barzilay, de Delta Electronics hoogleraar Elektrotechniek en Informatica; en Tommi Jaakkola, de Thomas Siebel hoogleraar Elektrotechniek en Informatica.
Lokaal optreden
Een enkel organisch molecuul kan uit tientallen en zelfs honderden atomen bestaan. Maar een reactie tussen twee van dergelijke moleculen kan slechts twee of drie atomen omvatten, die hun bestaande chemische bindingen verbreken en nieuwe vormen. Duizenden reacties tussen honderden verschillende reagentia komen vaak neer op een enkele, gedeelde reactie tussen hetzelfde paar 'reactiesites'.
Een groot organisch molecuul, echter, kan meerdere reactieplaatsen hebben, en wanneer het een ander groot organisch molecuul ontmoet, slechts één van de verschillende mogelijke reacties tussen hen zal daadwerkelijk plaatsvinden. Dit is wat automatische reactievoorspelling zo lastig maakt.
Vroeger, scheikundigen hebben computermodellen gebouwd die reacties karakteriseren in termen van interacties op reactieplaatsen. Maar ze vereisen vaak de opsomming van uitzonderingen, die onafhankelijk moeten worden onderzocht en met de hand moeten worden gecodeerd. Het model zou kunnen verklaren, bijvoorbeeld, dat als molecuul A reactieplaats X heeft, en molecuul B heeft reactieplaats Y, dan zullen X en Y reageren om groep Z te vormen - tenzij molecuul A ook reactieplaatsen P heeft, Q, R, S, T, jij, of V.
Het is niet ongebruikelijk dat een enkel model meer dan een dozijn opgesomde uitzonderingen vereist. En deze uitzonderingen in de wetenschappelijke literatuur ontdekken en toevoegen aan de modellen is een moeizame taak, die het nut van de modellen heeft beperkt.
Een van de belangrijkste doelen van het nieuwe systeem van de MIT-onderzoekers is om dit moeizame proces te omzeilen. Coley en zijn co-auteurs begonnen met 15, 000 empirisch waargenomen reacties gerapporteerd in Amerikaanse octrooiaanvragen. Echter, omdat het machine-leersysteem moest leren welke reacties niet zouden optreden, evenals degenen die dat zouden doen, voorbeelden van succesvolle reacties waren niet genoeg.
Negatieve voorbeelden
Dus voor elk paar moleculen in een van de vermelde reacties, Coley genereerde ook een batterij van aanvullende mogelijke producten, gebaseerd op de reactieplaatsen van de moleculen. Hij gaf vervolgens beschrijvingen van reacties, samen met zijn kunstmatig uitgebreide lijsten van mogelijke producten, naar een artificieel intelligentiesysteem dat bekend staat als een neuraal netwerk, die werd belast met het rangschikken van de mogelijke producten in volgorde van waarschijnlijkheid.
Van deze opleiding het netwerk leerde in wezen een hiërarchie van reacties - welke interacties op welke reactiesites de neiging hebben voorrang te krijgen op welke andere - zonder de moeizame menselijke annotatie.
Andere kenmerken van een molecuul kunnen de reactiviteit beïnvloeden. De atomen op een bepaalde reactieplaats kunnen, bijvoorbeeld, verschillende ladingsverdelingen hebben, afhankelijk van welke andere atomen zich om hen heen bevinden. En de fysieke vorm van een molecuul kan een reactieplaats moeilijk toegankelijk maken. Het model van de MIT-onderzoekers bevat dus ook numerieke metingen van beide kenmerken.
Volgens Richard Robinson, een onderzoeker chemische technologie bij het geneesmiddelenbedrijf Novartis, het MIT-onderzoekerssysteem "biedt een andere benadering van machine learning op het gebied van gerichte synthese, die in de toekomst de praktijk van experimenteel ontwerp zou kunnen transformeren naar gerichte moleculen."
"Momenteel leunen we sterk op onze eigen retrosynthetische training, die is afgestemd op onze eigen persoonlijke ervaringen en is aangevuld met zoekmachines in de reactiedatabase, Robinson zegt. "Dit komt ons goed van pas, maar resulteert vaak nog steeds in een aanzienlijk percentage mislukkingen. Zelfs zeer ervaren chemici zijn vaak verrast. Als je als industrie alle cumulatieve synthesefouten zou optellen, dit zou waarschijnlijk gepaard gaan met een aanzienlijke tijd- en kosteninvestering. Wat als we ons slagingspercentage konden verbeteren?"
De MIT-onderzoekers, Robinson zegt, "hebben op slimme wijze een nieuwe benadering aangetoond om hogere voorspellende reactieprestaties te bereiken dan conventionele benaderingen. Door de gerapporteerde literatuur aan te vullen met voorbeelden van negatieve reacties, de dataset heeft meer waarde."
Dit verhaal is opnieuw gepubliceerd met dank aan MIT News (web.mit.edu/newsoffice/), een populaire site met nieuws over MIT-onderzoek, innovatie en onderwijs.
 Timing van grote aardbevingen volgt een trappatroon van duivels
Timing van grote aardbevingen volgt een trappatroon van duivels- Boeren Vanuatu vulkaan kan blazen, krachten 7, 000 om te vluchten
- Duizenden protesteren tegen door China gesteunde megadam in Myanmar
- Kelps-recordreis stelt Antarctische ecosystemen bloot aan verandering
- Mexico's weg naar herstel na aardbevingen is veel langer dan het lijkt
Hoofdlijnen
- Maak kennis met de kleine machines in cellen die virussen afslachten
- Garnalenvisserij in New England voor minstens een jaar gesloten
- Zijn de hersenen bedraad voor religie?
- Is de Krebs-cyclus aëroob of anaëroob?
- Duiven beter in multitasken dan mensen:studie
- Een door kracht aangedreven mechanisme voor het vaststellen van celpolariteit
- Waarom zijn er 61 Anticodonen?
- Wat is de functie van een eicel?
- Verander je geliefde in een boom met Bios Urn
- Nieuwe verbinding kan voorkomen dat bacteriën ziekte veroorzaken

- Chemici suggereren een nieuwe manier om steroïde-analogen te synthetiseren
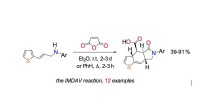
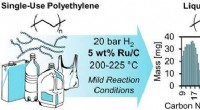
- Een milde manier om plastic in flessen te upcyclen tot brandstof en andere hoogwaardige producten
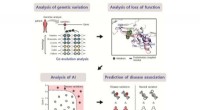
- Evolutionaire koppelingsanalyse identificeert de impact van ziektegerelateerde varianten
- Een beschermend schild voor gevoelige enzymen in biobrandstofcellen


 Het verschil tussen korte en lange termijn geheugen
Het verschil tussen korte en lange termijn geheugen- Beeldvorming van explosieve ontstekers
- Nieuwe dinosaurussoort ontdekt op Isle of Wight
- Jets van zwarte gaten kunnen de stervorming in sterrenstelsels beïnvloeden door interstellair gas te verspreiden en te verwarmen
- Hebben vogelgezang en menselijke spraak biologische wortels?
- Rewilding:zeldzame vogels keren terug als het grazen van vee is gestopt
- Mensen versus automatisering:agenten van servicecentra kunnen beter presteren dan technologie, studie toont
- Waarom het studeren van kunst zoals acteren of dans zakelijke studenten beter kan toerusten voor de post-COVID-wereld
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
