Wetenschap
Evolutionaire koppelingsanalyse identificeert de impact van ziektegerelateerde varianten
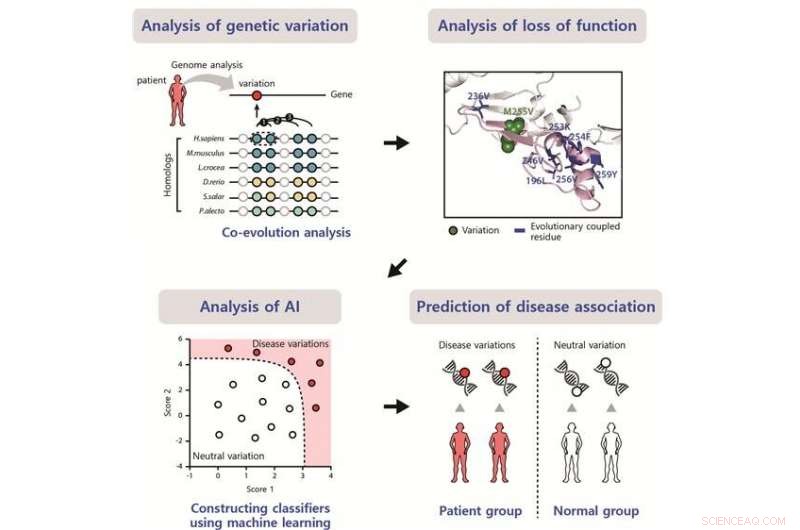
Schema van de ontwikkelde methode om de impact van ziektegerelateerde varianten te identificeren. Krediet:POSTECH
Het voorspellen van de impact van DNA-sequentievarianten is belangrijk voor het sorteren van ziektegerelateerde varianten (DV's) van neutrale varianten. Koreaanse onderzoekers van Pohang University of science and technology (POSTECH) rapporteren de ontwikkeling van een methode om de impact van DV's te voorspellen. De studie verschijnt in het tijdschrift Onderzoek naar nucleïnezuren in juni.
De huidige methoden om de mutatie-effecten te voorspellen, zijn afhankelijk van evolutionair behoud op de mutatieplaats, die wordt bepaald met behulp van homologe sequenties en gebaseerd op de veronderstelling dat varianten op goed geconserveerde locaties een hoge impact hebben. Echter, veel DV's op minder geconserveerde maar functioneel belangrijke locaties kunnen met de huidige methoden niet worden voorspeld.
De onderzoekers presenteren een methode om DV's te vinden op minder geconserveerde locaties door de mutatie-effecten te voorspellen met behulp van evolutionaire koppelingsanalyse. Functioneel belangrijke en evolutionair gekoppelde sites hebben vaak compenserende varianten op coöperatieve sites om functieverlies te voorkomen. Ze identificeerden DV's op minder geconserveerde locaties die niet werden geïdentificeerd met behulp van de huidige op natuurbehoud gebaseerde methoden.
Prof. Kim zei, "Deze studie kan worden toegepast op een verscheidenheid aan benaderingen van precisiegeneeskunde, zoals de prognose van de ziekten van de patiënt en het vinden van gepersonaliseerde geneeskunde. Gebaseerd op een grootschalige sequentieanalyse, de ontwikkelde methode is nuttig om meer ziektegerelateerde varianten te vinden die helpen bij het vinden van biomarkers en therapeutische doelen van verschillende menselijke ziekten."
 Ontdekking binnen het celcyclusproces om inzicht te krijgen in cellulaire ziekten
Ontdekking binnen het celcyclusproces om inzicht te krijgen in cellulaire ziekten- Poreuze koolstofnanovezels vertonen uitzonderlijke capacitieve deïonisatie
- Moleculaire engineering metaalcoördinatie-interacties voor sterke, moeilijk, snel herstellende hydrogels
- Guardians of the Ring - Onderzoekers onthullen de structuur van eiwitten die betrokken zijn bij ontstekingsziekten
- Veilig printen met onzichtbare inkt op waterbasis
- Welke vogels zijn pinguïns die het meest verwant zijn met?
- Plastic microvezels voor het eerst gevonden in ontlasting van wilde dieren van Zuid-Amerikaanse pelsrobben
- Vulkaanuitbarstingen hebben meer effect in de zomer
- Hoge niveaus van kankerverwekkende chemische stoffen gevonden in alledaagse consumentenproducten
- Menselijke invloed op klimaatverandering zal leiden tot meer extreme hittegolven in de VS
Hoofdlijnen
- Vijf stappen om Agar te bereiden Slants
- Studie identificeert waarschijnlijke scenario's voor wereldwijde verspreiding van verwoestende gewasziekte
- Wat is het doel van de fibreuze capsule?
- Vissen geven inzicht in de evolutie van het immuunsysteem
- Kan glucose door het celmembraan diffunderen door eenvoudige diffusie?
- Waarom is de studie van de histologie belangrijk in uw algehele begrip van anatomie en fysiologie?
- Hoe krijgen mensen zuurstof in hun lichaam?
- Het hormoon dat uw hond agressief zou kunnen maken
- Betere mensen maken - Het huwelijk tussen mens en machine
- Drie overeenkomsten tussen een verbinding en een element

- Nieuw onderzoek laat zien hoe geclusterde deeltjes de elasticiteit van sommige gels bepalen

- Haaibestendig materiaal voor wetsuits kan levens helpen redden

- Flexibele elektronische huid helpt mens-machine-interacties

- Bijna 200 jaar geleden ontdekte kristalstructuur zou de sleutel kunnen zijn tot zonnecelrevolutie


 Animal News Roundup! Drie rare nieuwe ontdekkingen die je moet weten
Animal News Roundup! Drie rare nieuwe ontdekkingen die je moet weten - Studie onthult nieuwe structuur van goud bij extremen
- Graduate research fellow onderzoekt hoe schimmels en vuur het ecosysteem van dennensavannes laten gedijen
- Hoe beïnvloedt de omwenteling van de aarde zijn seizoenen?
- De code van de Dode Zeerollen kraken
- Onderzoekers creëren transparante, rekbare geleiders met nano-accordeonstructuur
- Waarom is de Steenbokskeerkring belangrijk?
- Bronstijd Scandinavië handelsnetwerken voor koper geregeld
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
