Wetenschap
Een betere manier om RNA-virusnaalden te vinden in hooibergen van databases
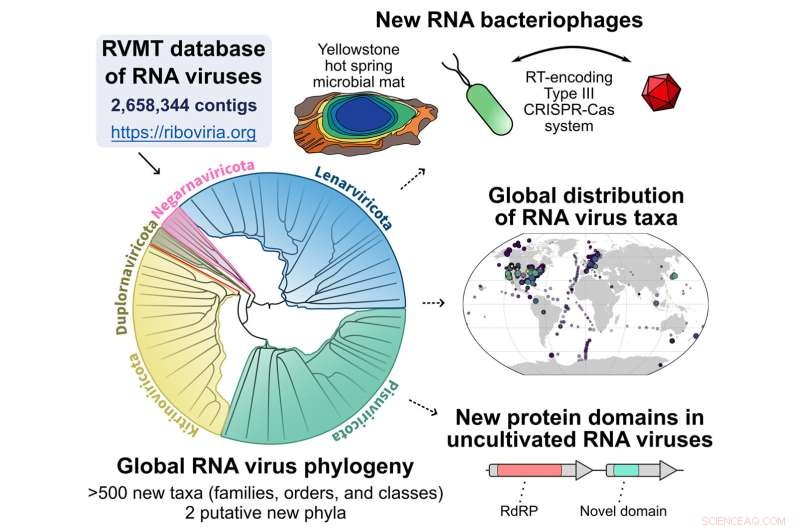
Grafisch overzicht van de pijplijn die begint met de RNA Virus MetaTranscriptomes (RVMT)-database om de uitbreiding van de diversiteit van RNA-virussen aan het licht te brengen. Krediet:Simon Roux
Een dierentuin bood ooit een kleurboek aan met ijsberen in wintertaferelen met kleurpotloden in verschillende tinten wit. Voor onderzoekers die op zoek zijn naar sequenties van RNA-virussen in grote datasets, kan hun werk lijken op het vinden van een enkele sneeuwvlok op een ingekleurde pagina van dat boek.
Online gepubliceerd op 28 september 2022 in Cell , een team onder leiding van onderzoekers van de Universiteit van Tel Aviv in Israël, het National Center for Biotechnology Information en het Joint Genome Institute (JGI) van het Amerikaanse Department of Energy (DOE), een DOE Office of Science User Facility in het Lawrence Berkeley National Laboratory ( Berkeley Lab) beschrijven een computationele pijplijn die specifiek kan scannen op die sneeuwvlokken of RNA-virussequenties. Met behulp van deze workflow heeft het team meer dan 5.000 datasets van RNA-sequenties (metatranscriptomen) uitgekamd die zijn gegenereerd uit verschillende omgevingsmonsters over de hele wereld, wat resulteerde in een vijfvoudige toename van de diversiteit van RNA-virussen.
"De wereld van virussen om ons heen is enorm, en we hebben nu de middelen om het te verkennen", zegt Eugene Koonin, een senior onderzoeker bij de NCBI en een van de senior auteurs van de krant, over de ontdekte virale diversiteit. "Hoewel de technische uitdagingen van data-analyse op deze schaal formidabel zijn."
Computationele zeven om reeksen te filteren
Er zijn meer microben op de planeet dan deeltjes in een handvol vuil, en virussen zijn veel groter dan de microben. Vooruitgang in sequencing-technologieën en computerhulpmiddelen heeft een diversiteit aan virussen blootgelegd die niet alleen gewassen, dieren en mensen infecteren, maar ook microben waarvan de aan- of afwezigheid de voedingscycli van de planeet kan beïnvloeden.
Terwijl de genetische informatie van de meeste organismen is gecodeerd in DNA, waarbij RNA de instructies in DNA aan de cel levert, slaan RNA-virussen hun genetische informatie op in RNA zonder een DNA-stadium. "Ik zou beweren dat RNA-virussen wereldwijd zelfs minder bekend zijn dan DNA-virussen", zegt Simon Roux, een JGI-wetenschapper en een van de mede-leiders van het project. "Maar net als DNA-virussen infecteren RNA-virussen microben over de hele wereld en leiden ze tijdens infectie tot celdood en/of ingrijpende veranderingen in de celfysiologie."
Hoewel alle RNA-virussen een gen hebben dat codeert voor een enzym genaamd RNS-directed RNA Polymerase (RdRP), dat nodig is voor het repliceren van de RNA-genoomreplicatie, was het een uitdaging om het te detecteren. Het vinden van de RNA-virussneeuwvlokken in de sneeuwstorm van genomische gegevens omvatte de ontwikkeling van speciale rekenzeven om sequenties uit te filteren waarvan het onwaarschijnlijk was dat ze de RdRP-sequentie bevatten.
Het werk was het resultaat van een samenwerking in drie richtingen die begon in 2019, herinnert Uri Neri van de Universiteit van Tel Aviv, een van de co-leads van het project en eerste auteur van de studie. Leden van de teams van Tel Aviv en NCBI, die al samen bezig waren met het delven van prokaryotische virussen, hoorden van Nikos Kyrpides van JGI dat zijn Microbiome Data Science-groep ook bezig was met het delven van RNA-virussen. Na een paar virtuele bijeenkomsten van de drie teams was het duidelijk dat een grotere gezamenlijke inspanning veel effectiever zou zijn in het bereiken van resultaten van hogere kwaliteit in vergelijking met kleinere individuele inspanningen. Dit is ook het soort synergetische en collaboratieve gemeenschapszin waar het JGI voor pleit en actief promoot.
Het team gebruikte alle openbaar beschikbare metatranscriptoomdatasets van het JGI's Integrated Microbial Genomes &Microbiomes (IMG/M)-systeem. "Vervolgens hebben we veel meer monsters onderzocht en onze methodologie verfijnd", zei Neri. "Ons team groeide en de omvang van het project ook." Hiertoe, benadrukte Kyrpides, kunnen de bijdragen van de talrijke wetenschappelijke gebruikers van JGI bij het verzamelen en indienen van hun microbioommonsters voor sequencing bij het JGI niet worden overschat. Hun medewerking en steun, zei hij, en in verschillende gevallen hun toestemming om nog niet-gepubliceerde sequentiegegevens te gebruiken, was absoluut cruciaal voor het succes van deze inspanning en dat gold ook voor de erkenning van hun bijdrage.
Zowel Roux als Koonin merkten op dat de overvloed aan ontdekte RNA-virussequenties "het globale beeld van virusdiversiteit aanzienlijk verandert", hoewel niet op de hogere classificaties van virusgroepen (phyla). groepen terwijl ook nieuwe takken worden toegevoegd. Bovendien lijken RNA-virussen niet gelijkmatig over de wereld te zijn verspreid.
Een uitgebreide groep is van virussen geassocieerd met bacteriën; tot nu toe zijn de meeste bekende RNA-virussen in verband gebracht met eukaryoten. Samen met de uitbreiding van met bacteriën geassocieerde RNA-virussen is de bevinding dat "een paar bacteriën CRISPR gebruiken om zich tegen RNA te verdedigen", merkte Roux op, "hoewel het onduidelijk is waarom dit zo zelden wordt gedetecteerd."
Ontwikkelen van benaderingen voor het verzoenen van 'echte' Big Data
Voor het team is het computationele werk dat leidde tot de ontdekte overvloed aan RNA-virussen nog maar het begin. "Ik zeg vaak dat alleen het identificeren van een sequentie als viraal nog niet het halve verhaal is." zei Neri. "We hebben veel van onze inspanningen geïnvesteerd in de analyses na de ontdekking - we hebben zo goed mogelijk geprobeerd de eiwitdomeinen te beschrijven die elk virus draagt, en wie hun waarschijnlijke gastheer is. We hebben al die informatie volledig gratis en openlijk gemaakt beschikbaar voor de bredere wetenschappelijke gemeenschap."
Uri Gophna van de Universiteit van Tel Aviv en Koonin merkten allebei op dat ander parallel onderzoek vergelijkbare "dramatische uitbreidingen" van het wereldwijde RNA-viroom heeft gemeld. "We moeten nu de bevindingen vergelijken en met elkaar in overeenstemming brengen, en komen tot een enkele, niet-redundante dataset", zei Koonin. "Hopelijk kunnen we relatief snel de werkelijke grootte van het RNA-viroom schatten. Dit is nu echter echte Big Data, we hebben te maken met miljarden sequenties en binnenkort met biljoenen. De ontwikkeling van efficiënte, geautomatiseerde analysemethoden en het classificeren van sequentiegegevens op deze schaal is essentieel." + Verder verkennen
Een geautomatiseerd hulpmiddel voor het beoordelen van de kwaliteit van virusgegevens
 Vereenvoudigde methode maakt celvrije eiwitsynthese flexibeler en toegankelijker
Vereenvoudigde methode maakt celvrije eiwitsynthese flexibeler en toegankelijker- De zuurtest doorstaan:nieuw systeem met lage pH recyclet meer koolstof in waardevolle producten
- Het nabootsen van termieten om nieuwe materialen te genereren
- Hoe worden oxidatiereductiereacties gebruikt in het dagelijks leven?
- Algoritme voor machinaal leren helpt bij het zoeken naar nieuwe medicijnen
- Aardbevingen schudden de watersystemen van Nieuw-Zeeland op z'n kop
- Geofysicus:gewicht van Harvey-regens deed Houston zinken
- Methoden voor verwijdering van huishoudelijk afval
- Voorkomen van toename van extreme hitte in Oost-Azië met 0,5 graden C
- Onderzoek toont aan dat planten in Afrika groen worden voor het regenseizoen
Hoofdlijnen
- Het gedrag van primaten veranderde toen dierentuinen gesloten waren wegens pandemie, suggereert onderzoek
- Nadelen en voordelen van een HPLC
- Een botanisch mysterie opgelost door fylogenetische tests
- Waarschijnlijkheden in de genetica: waarom is het belangrijk?
- De allereerste tagging van Amazone-dolfijnen om de inspanningen voor natuurbehoud te stimuleren
- Isle Royale wolvenpopulatie stijgt nadat ze bijna is uitgestorven
- Plantenveredeling:onzichtbare chromosomen gebruiken om pakketjes positieve eigenschappen door te geven
- Deskundigen zijn het erover eens:het welzijn van melkvee is slechter dan vleesvee
- Lijst met celorganellen en hun functies
- Vier nieuwe caladiumcultivars voor containers en landschappen

- Urban Coopers-haviken overtreffen hun landelijke buren

- Onderzoek suggereert dat gevaarlijke gewassenschimmel giftige chemicaliën produceert om insecten af te weren
- Onderzoekers identificeren een paar receptoren die essentieel zijn voor de communicatie tussen mannen en vrouwen bij planten
- Door te bezuinigen op milieu-uitgaven, de overheid snijdt kansen


 Stellaire wind van oude sterren onthult het bestaan van een partner
Stellaire wind van oude sterren onthult het bestaan van een partner- Observatorium op grote hoogte werpt licht op de oorsprong van overtollige antimaterie
- Praktisch gebruik van Distillation
- Wetenschappers bestuderen atmosferische golven die uit orkanen komen
- Moleculaire motoren:Chemische carrousel draait in de kou
- InSight-lander bij de nieuwste ExoMars-afbeeldingspremie
- Licht uit in Sydney voor behoudscampagne Earth Hour
- Hubble ziet poollicht op Uranus
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
