Wetenschap
Genomics onthult hoe concurrentie tussen bacteriën de impact van vaccinatie beïnvloedt
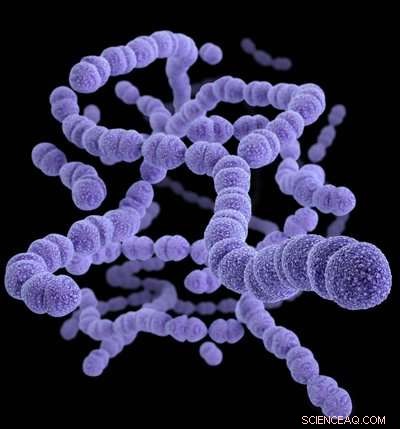
Streptococcus pneumoniae. Krediet:CDC/James Archer
Een grootschalig genetisch en modelleringsonderzoek van Streptococcus pneumoniae heeft nieuw inzicht opgeleverd in hoe recent geïntroduceerde vaccins veel stammen van de soort hebben geëlimineerd, en de verschillende manieren waarop de resterende bacteriën strijden om de kans om ze te vervangen.
S. pneumoniae wordt meestal gevonden achter de achterkant van de neusholte, waar het normaal gesproken onschadelijk is. Echter, het kan naar andere lichaamssites gaan, resulterend in het veroorzaken van duizenden gevallen van ernstige pneumokokkenziekte per jaar in het VK, en een veel grotere ziektelast in veel lage- of middeninkomenslanden. Deze infecties leiden tot longontsteking, bloedbaaninfecties of meningitis, en komen het meest voor bij jonge zuigelingen en ouderen. In antwoord, in het VK zijn twee verschillende vaccins geïntroduceerd om S. pneumoniae te bestrijden:het 7-valente vaccin in 2006, vervangen door het 13-valent vaccin in 2010. Sinds kinderen het vaccin kregen, de incidentie van pneumokokkenziekte is gedaald.
Na een paar jaar routinematige vaccinatie zijn veel stammen van S. pneumoniae, waaronder de meest voorkomende ziekteveroorzakende, was geëlimineerd. Nog, de bacteriën zijn niet minder gewoon geworden in hun ongevaarlijke habitat achter in de neusholte. In plaats daarvan waren de op het vaccin gerichte stammen vervangen door andere die minder vaak ziekten bij kinderen veroorzaken. Het laatste onderzoek, gepubliceerd in Natuur Ecologie &Evolutie , geeft een nieuwe verklaring voor hoe dit kan gebeuren.
Deze studie gebruikte drie grote verzamelingen genomen, gesequenced bij het Wellcome Trust Sanger Institute, om de effecten van vaccinatie in het VK te volgen, VS, en Nederland. In plaats van te kijken naar de verandering in de prevalentie van de individuele stammen zelf, de onderzoekers keken naar de frequenties van genen binnen de zeer diverse S. pneumoniae-populatie.
Terwijl verschillende S. pneumoniae-stammen domineren op verschillende locaties, de gedetailleerde informatie die beschikbaar is via sequencing van het hele genoom onthulde dat elke S. pneumoniae-populatie vergelijkbaar was in termen van genfrequenties. Dit was ook het geval bij het vergelijken van bacteriepopulaties aan het begin van de vaccinatieprogramma's met dezelfde locaties een paar jaar later. De computationele modellering van de onderzoekers toonde aan dat dit hoogst onwaarschijnlijk was dat dit door toeval was gebeurd.
Om de dynamiek van genfrequenties nauwkeurig te onderzoeken, de onderzoekers deden een beroep op de rijkdom aan genetische data die recentelijk is verzameld en op nieuwe wiskundige methoden om computersimulaties te vergelijken met actuele data. Het nieuwe algoritme ontwikkeld door Jukka Corander van het Infection Genomics-team van het Sanger Institute maakte nauwkeurigere modellering mogelijk en versnelde de snelheidsresultaten met behulp van dergelijke modellen tot 10, 000-voudig.
De resultaten suggereren het belang van een bepaald type natuurlijke selectie, waarin genen voordeliger zijn voor de bacteriën waarin ze worden gevonden als ze zeldzamer zijn - vandaar de naam, 'negatieve frequentieafhankelijke selectie'. Dit kan het gevolg zijn van concurrentie tussen bacteriestammen, en gedetailleerde analyse van de genoomsequenties suggereert de vele manieren waarop dit kan gebeuren. Bijvoorbeeld, S. pneumoniae-cellen scheiden chemicaliën af om de groei van andere stammen te voorkomen, vatbaar zijn voor infectie door verschillende virussen, verschillende strategieën toepassen voor het verkrijgen van voedingsstoffen, en verschillen in hoe ze worden herkend door het menselijke immuunsysteem. Het relatieve belang van deze verschillende processen wordt nog steeds niet goed begrepen.
Dit model belooft nieuw licht te werpen op waarom sommige bacteriën zulke complexe populatiestructuren hebben die het moeilijk maken om ze volledig te elimineren door vaccinatie. De auteurs vermoeden dat de negatieve frequentieafhankelijke selectie die plaatsvindt op genniveau een algemeen mechanisme is bij verschillende bacteriesoorten. Naarmate het gebruik van sequencing van het gehele genoom voor epidemiologische surveillance routinematiger wordt, dergelijke studies zullen een waardevolle basis vormen voor zowel het ontwerpen van betere controlestrategieën, en voor een beter begrip van hun nawerkingen.
 Overeenkomsten tussen geleiders en isolatoren
Overeenkomsten tussen geleiders en isolatoren - Onderzoekers ontrafelen de geheimen van hoe natuursteen gloeit in het donker
- Terahertz-golven onthullen verborgen processen in ultrasnelle kunstmatige fotosynthese
- Nieuwe verbinding effectief tegen medicijnresistente pathogenen, kan leiden tot nieuwe antibiotica
- Afbreekbare elektronische componenten gemaakt van maïszetmeel
- Waarom een koolstofprijs alleen niet genoeg zal zijn om de uitstoot van Nieuw-Zeeland te verlagen?
- 8 Ontzagwekkende feiten over de Stille Oceaan
- Gezelschapsdieren in de klimaatcrisis:hoe eigenaren van gezelschapsdieren hun impact op het milieu kunnen verminderen
- Hittegolf Zuid-Australië breekt recordtemperaturen
- NASA's GPM toont regenval ten zuidoosten van de afgescheurde tropische cycloon Iris
Hoofdlijnen
- Wat is een interne regulator van de celcyclus?
- Welke moleculen kunnen er zonder hulp door het plasmamembraan passeren?
- Baanbrekende nieuwe technologie om de wereldwijde zoektocht naar gewasverbetering te versnellen
- Hoge prijzen van dierlijke producten onderdeel van een vicieuze cirkel naar uitsterven
- Hoe slapende listeria zich in cellen verbergt
- Hoe de gecorrigeerde WBC-telling te berekenen
- Hoe afnemende zoogdierpopulaties in de Florida Everglades verband houden met de invasieve Birmese python
- Hoe beïnvloeden je hersenen je overlevingskansen in de wildernis?
- DropSynth, een eenpotsbenadering van gensynthese
- Wat zijn drie dingen die bepalen of een molecuul in staat is om door een celmembraan te diffunderen?

- DNA-onderzoek in de Stille Oceaan onthult een toename van 2000 procent in onze kennis van de biodiversiteit van weekdieren
- Lysosoom: definitie, structuur en functie
- Biologen maken toolkit voor het afstemmen van genetische circuits
- Is er echt een kurkcrisis?


 Watervoorspelling is somber voor groot stuwmeer in het zuidwesten van de VS
Watervoorspelling is somber voor groot stuwmeer in het zuidwesten van de VS- Nieuw voorspellingsmodel zorgt voor het vroegste bewustzijn ooit van overstromingen en droogtes wereldwijd
- Facebook kan het aantal likes voor berichten verbergen
- Trump zegt dat NASA moet stoppen met praten over teruggaan naar de maan
- Stichtingsfinanciering verandert internationale berichtgeving
- Zeldzaam Isaac Newton-manuscript ontdekt in Corsicaanse bibliotheek
- Zwarte sneeuw valt op Siberië, toegeschreven aan moordende kolenindustrie
- Zijn ecobricks het antwoord op plasticvervuiling?
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
