Wetenschap
Hoe markergenen in celclusters te vinden
Grafisch abstract. Credit:Journal of Molecular Biology (2022). DOI:10.1016/j.jmb.2022.167525
Welke genen zijn specifiek voor een bepaald celtype, d.w.z. "markeren" hun identiteit? Met de toenemende omvang van datasets tegenwoordig, is het beantwoorden van deze vraag vaak een uitdaging. Vaak zijn markergenen gewoon genen die in specifieke celpopulaties zijn gevonden. Er kunnen echter veel meer genen kenmerkend zijn voor een bepaald celtype, maar blijven onontdekt.
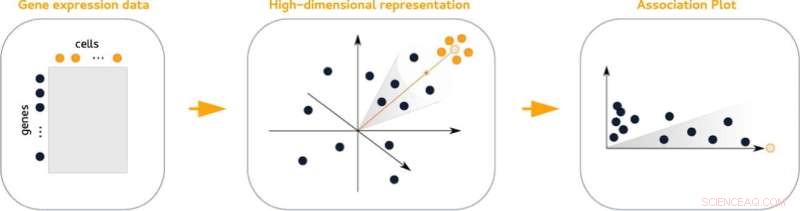
Associatieplots (APL), een nieuwe statistische methode voor het visualiseren van genactiviteit binnen een celcluster, maken het gemakkelijker om de markergenen te vinden. De plots vergelijken de activiteit van genen van een bepaald cluster met alle andere clusters uit de dataset. Bovendien maken ze het gemakkelijk om te zien welke genen worden gedeeld met andere clusters.
"Associatieplots stellen ons niet alleen in staat om nieuwe markergenen te identificeren. Het werkt ook andersom:we kunnen clusters van onbekende identiteit in een dataset koppelen aan celtypes, op basis van een verstrekte lijst met markergenen", zegt Elzbieta Gralinska. van het Max Planck Instituut voor Moleculaire Genetica in Berlijn.
De biotechnoloog werkt in het team van Martin Vingron, dat de techniek heeft ontwikkeld. De onderzoekers demonstreerden de functionaliteit van de techniek op twee openbaar beschikbare datasets en publiceerden de resultaten in het Journal of Molecular Biology . Bovendien is APL uitgebracht als een gratis module voor de statistische omgeving R. Met het APL-pakket kunnen onderzoekers hun eencellige gegevens visueel inspecteren en individuele genen selecteren met de cursor om meer diepgaande details te leren.
Afzonderlijke cellen analyseren en groeperen
Waarom is het überhaupt nodig om markergenen te identificeren? Moderne sequencing-technologieën zijn in staat om individuele RNA-moleculen in individuele cellen te ontcijferen. Van een bloedmonster kan bijvoorbeeld elke cel worden gescheiden en een monster van de RNA's van de cel kan worden gedecodeerd. Deze eencellige gegevens vertegenwoordigen de actieve genen die werden getranscribeerd in RNA-moleculen.
Het voordeel:in plaats van te puzzelen over tot welk celtype een bepaald RNA behoort, kan het worden herleid tot de cel van oorsprong. Het nadeel:het sequencen van duizenden RNA's in elke afzonderlijke cel van tienduizenden cellen levert buitengewone hoeveelheden gegevens op.
Een uitweg is om de cellen te sorteren op basis van hun RNA-gehalte. "Eencellige gegevens zijn samengesteld uit een wilde mix van veel verschillende celtypen. We zijn geïnteresseerd in cellen van hetzelfde celtype, die zich allemaal hetzelfde zouden moeten gedragen", legt Martin Vingron uit. Daarom is het logisch om vergelijkbare cellen computationeel te groeperen, zegt hij. "Voor ons definiëren de markergenen een celtype."
Celclusters interactief verkennen
Met behulp van openbaar beschikbare gegevens van witte bloedcellen demonstreerde het team hoe het nieuwe algoritme werkt. De vele verschillende soorten witte bloedcellen zoals T-cellen, B-cellen of monocyten zijn allemaal gegroepeerd in afzonderlijke clusters. De onderzoekers bevestigden bekende markergenen en konden aantonen dat naaste verwanten tussen de bloedcellen ook grote overeenkomsten vertonen in hun genactiviteit.
"Elk van de markergenen die we met APL vonden, had kunnen worden ontdekt door ten minste één andere bestaande methode voor identificatie van markergenen", zegt Gralinska. Maar het voordeel van APL ten opzichte van de bestaande algoritmen is de grafische weergave van de resultaten, zegt ze. "Bestaande tools bieden lange lijsten met genen en scorewaarden. Vaak doorlopen gebruikers de lijst en stoppen ze bij een willekeurige grens."
De nieuwe methode biedt daarentegen een manier om deze genen te visualiseren, op elke genen te klikken en de activiteit ervan nader te bekijken, zegt ze. "We bieden niet alleen lijsten met markergenen, we laten gebruikers beoordelen hoe deze genen zich gedragen", zegt de onderzoeker. "Met associatieplots kunnen ze in hun gegevens duiken om meer te weten te komen over elk celtype." Bovendien, zegt ze, is het heel gemakkelijk om de biologische rol van de meest interessante genen in een volgende stap af te breken via verrijkingsanalyse van Gene Ontology-termen, die compatibel is met de APL-software - iets wat ze als "een zeer nuttige functie" beschouwt.
Het onderliggende wiskundige model
De hoogdimensionale gegevens die informatie bevatten over activiteit tussen genen kunnen niet visueel worden weergegeven zonder verlies van informatie. Hetzelfde geldt voor geclusterde gegevens, die de analyse bemoeilijken. "Onze truc is dat we met veel meer dan alleen twee of drie dimensies rekening houden, maar uiteindelijk een tweedimensionaal diagram maken", zegt Gralinska.
De associatiegrafieken zijn afgeleid van een wiskundige techniek die zowel genen als cellen tegelijkertijd in een gemeenschappelijke, hoogdimensionale ruimte insluit. Het meten van de afstanden tussen genen en een bepaald celcluster in deze ruimte resulteert in waardeparen die de associatie van een gen met een bepaald cluster weerspiegelen en inzicht geven in de associatie met andere clusters.
"Een tekortkoming van APL is dat we vertrouwen op vooraf geclusterde gegevens, wat betekent dat we voor clustering op andere technieken moeten vertrouwen", zegt Martin Vingron. "Toch hopen we dat onze nieuwe methode veel nieuwe gebruikers zal vinden. We vinden dat een visueel en interactief proces gewoon een betere analyse maakt."
 Synthese van waardevolle chemicaliën uit verontreinigde grond
Synthese van waardevolle chemicaliën uit verontreinigde grond- De verschillen in covalente kristallen en moleculaire kristallen
- Review benadrukt de kracht van eenvoudige fysieke modellen voor complexe eiwitmachines
- Fluorescerende techniek brengt verouderende polymeren aan het licht
- Wat gebeurt er met een kooktemperatuur als de druk daalt?
- NASA ziet tropische storm Karina's nacht bewegen
- Natuurlijke kleurstoftuin bevordert een groenere toeleveringsketen voor mode
- Afgedankte persoonlijke beschermingsmiddelen kunnen worden omgezet in biobrandstof, studie toont
- Mountain Time Vs. Pacific Time
- NASA ziet de wind uit de tropische storm worden geslagen John
Hoofdlijnen
- De vondst van zijn leven:8-jarige jongen ontdekt gigantische haaientand in South Carolina
- Wat is een regeling in de microbiologie?
- Internationale samenwerking werkt om schimmelresistent katoen te kweken
- Twee neuropeptiden in zebravissen geven aanwijzingen voor de complexe neurale mechanismen die ten grondslag liggen aan slaap
- Lawaai van industriële ontwikkeling zal dieren benadrukken en de ecologie in nationale monumenten veranderen
- Cross-species cellandschap gebouwd op eencellig niveau
- Hoe de cellen van planten, dieren en eencellige organismen te vergelijken
- Waarom zijn cellen belangrijk voor levende organismen?
- Het risico op een keizersnede is erfelijk:natuurlijke selectie kan het aantal belemmerde bevallingen niet verminderen
- Grootste database over zoogdieren in Portugal nu beschikbaar

- Jeugddieet kan bijdragen aan aangetaste verstandskiezen, blijkt uit onderzoek

- De verschillen tussen monosachariden en polysachariden

- Science Fair Project Ideas: Equine

- Pakken of niet aaien? Hoe de interactie tussen kinderen en honden veilig te houden


 Slowakije voelt de meeste pijn van Trump-autotarieven:experts
Slowakije voelt de meeste pijn van Trump-autotarieven:experts- Chimpansees brengen insecten aan op wonden, een mogelijk geval van medicijngebruik?
- Studie onderzoekt hoe instandhoudingsinitiatieven besmettelijker kunnen worden gemaakt
- In de wereld van schoonheidswedstrijden van een miljoen dollar - voor kamelen
- Republikeinse en Democratische kiezers zijn het over één ding eens:de noodzaak van genereuze COVID-19-hulp
- Portugal worstelt om uit de as van zijn dodelijkste vuur te herrijzen
- Sewall Green Wright
- Waarom is de trekduif uitgestorven?
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
