Wetenschap
De ontdekking van magneten met één molecuul versnellen met deep learning
Het synthetiseren of bestuderen van bepaalde materialen in een laboratoriumomgeving brengt vaak uitdagingen met zich mee vanwege veiligheidsproblemen, onpraktische experimentele omstandigheden of kostenbeperkingen. Als reactie daarop wenden wetenschappers zich steeds meer tot deep learning-methoden, waarbij machine learning-modellen worden ontwikkeld en getraind om patronen en relaties in gegevens te herkennen die informatie bevatten over materiaaleigenschappen, composities en gedrag.
Met behulp van deep learning kunnen wetenschappers snel voorspellingen doen over materiaaleigenschappen op basis van de samenstelling, structuur en andere relevante kenmerken van het materiaal, potentiële kandidaten voor verder onderzoek identificeren en de syntheseomstandigheden optimaliseren.
Nu blijkt uit een onderzoek dat verschijnt in International Union of Crystallography Journal (IUCrJ) Professor Takashiro Akitsu, assistent-professor Daisuke Nakane en de heer Yuji Takiguchi van de Tokyo University of Science (TUS) hebben deep learning gebruikt om single-molecule magneten (SMM's) te voorspellen uit een verzameling van 20.000 metaalcomplexen. Deze innovatieve strategie stroomlijnt het materiaalontdekkingsproces door de behoefte aan langdurige experimenten tot een minimum te beperken.
Single-molecule magneten (SMM's) zijn metaalcomplexen die magnetisch relaxatiegedrag demonstreren op individueel molecuulniveau, waarbij magnetische momenten in de loop van de tijd veranderingen of ontspanning ondergaan. Deze materialen hebben potentiële toepassingen in de ontwikkeling van geheugen met hoge dichtheid, kwantummoleculaire spintronische apparaten en kwantumcomputerapparatuur. SMM's worden gekenmerkt door een hoge effectieve energiebarrière (Ueff ) zodat het magnetische moment omdraait. Deze waarden liggen echter doorgaans in het bereik van tientallen tot honderden Kelvins, waardoor SMM's lastig te synthetiseren zijn.
De onderzoekers gebruikten deep learning om de relatie tussen moleculaire structuren en SMM-gedrag in metaalcomplexen met liganden van het salen-type te identificeren. Deze metaalcomplexen zijn gekozen omdat ze gemakkelijk kunnen worden gesynthetiseerd door aldehyden en aminen te complexeren met verschillende 3d- en 4f-metalen.
Voor de dataset hebben de onderzoekers tussen 2011 en 2021 uitgebreid gewerkt aan het screenen van 800 artikelen, waarbij ze informatie verzamelden over de kristalstructuur en bepaalden of deze complexen SMM-gedrag vertoonden. Bovendien verkregen ze structurele 3D-details van de moleculen uit de Cambridge Structural Database.
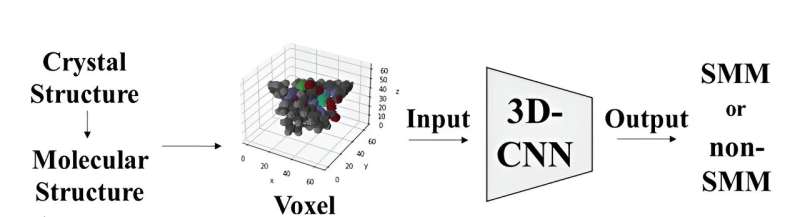
De moleculaire structuur van de complexen werd weergegeven met behulp van voxels of 3D-pixels, waarbij elk element een unieke RGB-waarde kreeg toegewezen. Vervolgens dienden deze voxelrepresentaties als input voor een 3D Convolutional Neural Network-model gebaseerd op de ResNet-architectuur. Dit model is specifiek ontworpen om moleculen te classificeren als SMM's of niet-SMM's door hun 3D-moleculaire beelden te analyseren.
Toen het model werd getraind op een dataset van kristalstructuren van metaalcomplexen die complexen van het salen-type bevatten, bereikte het een nauwkeurigheidspercentage van 70% bij het onderscheiden van de twee categorieën. Toen het model werd getest op 20.000 kristalstructuren van metaalcomplexen die Schiff-basen bevatten, ontdekte het met succes de metaalcomplexen die werden gerapporteerd als magneten met één molecuul.
"Dit is het eerste rapport over diepgaand leren over de moleculaire structuren van SMM's", zegt prof. Akitsu.
Bij veel van de voorspelde SMM-structuren waren multinucleaire dysprosiumcomplexen betrokken, bekend om hun hoge Ueff waarden. Hoewel deze methode het SMM-ontdekkingsproces vereenvoudigt, is het belangrijk op te merken dat de voorspellingen van het model uitsluitend gebaseerd zijn op trainingsgegevens en chemische structuren niet expliciet koppelen aan hun kwantumchemische berekeningen, een voorkeursmethode bij AI-ondersteund moleculair ontwerp. Verder experimenteel onderzoek is nodig om gegevens over SMM-gedrag onder uniforme omstandigheden te verkrijgen.
Deze vereenvoudigde aanpak heeft echter zijn voordelen. Het vermindert de behoefte aan complexe computerberekeningen en vermijdt de uitdagende taak van het simuleren van magnetisme.
Prof. Akitsu concludeert:"Het aannemen van een dergelijke aanpak kan richting geven aan het ontwerp van innovatieve moleculen, wat aanzienlijke besparingen in tijd, middelen en kosten kan opleveren bij de ontwikkeling van functionele materialen."
Meer informatie: Yuji Takiguchi et al, De voorspelling van magneeteigenschappen van één molecuul via deep learning, IUCrJ (2024). DOI:10.1107/S2052252524000770
Aangeboden door de Tokyo University of Science
 Synthese van middelgrote ringstructuren
Synthese van middelgrote ringstructuren- Onderzoekers ontdekken nieuwe transportroute voor vluchtige plantenstoffen
- Team bewijst het concept van een natuurlijke benadering van anti-transpiranten
- Raman- en infraroodspectroscopie helpen bij het identificeren van verschillende geacetyleerde lysines
- Waarom is gedistilleerd water een goede controle voor wetenschappelijke projecten?
- Het landschap bepaalt hoe beken hersteld moeten worden
- De wetenschap van koolstofdioxide en klimaat
- Hoe verspreiden vogels zaden?
- Antarctische ijsplaten:onderzoek onthult een ontbrekend stukje van de klimaatpuzzel
- Uit onderzoek blijkt dat het brandstofverbruik van een auto kan worden geannuleerd bij uw volgende aankoop van een auto
Hoofdlijnen
- Onderzoekers vinden dat RNA-bewerkingssites waarschijnlijk een belangrijkere rol spelen bij genetische ziekten
- Sporen van historische rendierweide zijn na 100 jaar nog steeds te zien
- Hoe is mitose van invloed op het leven?
- Stappen van DNA-transcriptie
- Wat zijn de primaire functies van fosfolipiden?
- Zijn domme mensen gelukkiger?
- Onderzoek toont aan dat honden slimmer zijn dan katten
- Hoe overtuigend is een Y-chromosoomprofielmatch tussen verdachte en plaats delict?
- Bereidheid om risico's te nemen - een persoonlijkheidskenmerk
- Onderzoekers zien in realtime scheurvorming in 3D-geprint wolfraam

- Wetenschappers ontwikkelen manier om salmonella-infectie in realtime te volgen
- Snelle en gemakkelijke synthese van antibacteriële aminozuur Schiff-base kopercomplexen
- Nieuwe goedkope en niet-toxische methode voor het maken van benzeenringen

- Eiwitstructuur zou nieuwe behandelingen voor cystische fibrose kunnen ontsluiten


 Driedimensionale puntenwolken geven inzicht in de structurele complexiteit van bossen
Driedimensionale puntenwolken geven inzicht in de structurele complexiteit van bossen- Wetenschappers zeggen dat een One Health-benadering van plantgezondheid van vitaal belang is voor het bereiken van duurzame wereldwijde voedselzekerheid
- Naar netto-energiezuivering van afvalwater met behulp van de huidige technologie
- Papieren strips kunnen toxines in drinkwater snel detecteren
- Experimentresultaten bevestigen anomalie, kunnen wijzen op nieuw elementair deeltje
- Nieuwe metingen tonen aan dat de maan gevaarlijke stralingsniveaus heeft
- De cirkel rond koolstofemissies van chemische fabrieken sluiten
- Kristallisatie kristalhelder gemaakt
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
