Wetenschap
Studie ontwikkelt nieuwe manier om kankercellen te identificeren
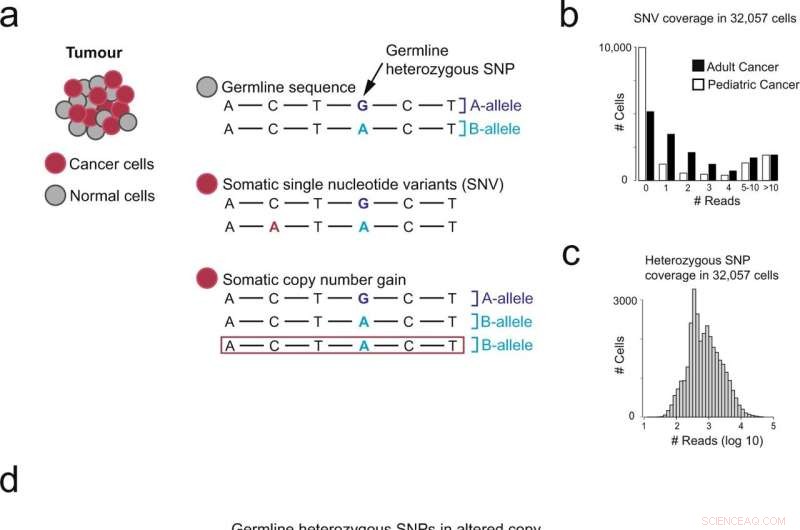
Overzicht van verschillende benaderingen voor het identificeren van van kanker afgeleide cellen. a Genomische veranderingen aanwezig in kankergenomen. b Aantal cellen (y-as) met N-waarden voor puntmutaties (x-as), gescheiden door lage (NB neuroblastoom) en hoge (RCC niercelcarcinoom) mutatiebelasting. c Aantal cellen (y-as) met N-waarden voor heterozygote single-nucleotide polymorfismen (SNP) (x-as). d Overzicht van het gebruik van allelische verschuivingen die veranderingen in het aantal kopieën vertegenwoordigen om kankercellen te detecteren. Krediet:Communicatiebiologie (2022). DOI:10.1038/s42003-022-03808-9. https://www.nature.com/articles/s42003-022-03808-9
Een nieuwe methode om kankercellen te scheiden van niet-kankercellen is ontwikkeld door onderzoekers van het Wellcome Sanger Institute, in een boost voor diegenen die kankerbiologie beter willen begrijpen met behulp van single-cell mRNA-sequencing.
De studie, vandaag gepubliceerd in Communications Biology , verbetert bestaande methoden om te identificeren welke cellen in een monster kankerachtig zijn en levert cruciale gegevens over de micro-omgeving van tumoren. De software is openlijk beschikbaar voor onderzoekers over de hele wereld om hun eigen gegevens toe te passen, waardoor de effectiviteit van eencellige sequencing om kanker beter te begrijpen, wordt verbeterd.
Eencellige mRNA-analyse van kankercellen is een van de meest geavanceerde technieken die worden gebruikt om kankerbiologie beter te begrijpen. De gegenereerde gegevens kunnen worden gebruikt om te proberen kankers te verstoren met medicijnen of om erachter te komen hoe kankers in de eerste plaats ontstaan.
Een fundamentele stap in dit proces is het scheiden van kankercellen en niet-kankercellen, maar dit is niet altijd een gemakkelijke taak. Naast de vele soorten kanker zullen er ook moleculaire verschillen zijn tussen kankercellen van hetzelfde type binnen een enkele tumor.
Momenteel is de beste methode om dit te doen het meten van de gemiddelde genexpressie van cellen in het monster, waarbij hogere of lagere expressie wordt gebruikt om kankercellen te onderscheiden van gezonde cellen. Maar deze methode kan tot verkeerde conclusies leiden.
In deze nieuwe studie voerden onderzoekers van het Wellcome Sanger Institute volledige genoomsequencing en single-cell mRNA-sequencing uit op monsters verzameld door Great Ormond Street Hospital (GOSH).
Door onevenwichtigheden van allelen in deze gegevens te lokaliseren, die wijzen op veranderingen in het aantal kopieën in het genoom, was het team in staat kankercellen betrouwbaarder te identificeren dan met eerdere methoden. Deze benadering zal vooral nuttig zijn voor het valideren van nieuwe kankerceltypes en een beter begrip van de micro-omgeving van tumorweefsel.
"In staat zijn om te weten hoe het transcriptoom anders is in cellen met afwijkende genomen, zoals die gevonden worden in kankers, is waardevolle kennis en zal de vragen uitbreiden die we kunnen beantwoorden met behulp van single-cell sequencing", zegt Dr. Matt Young.
De methode, allelIntegrator genaamd, is beschikbaar als softwarepakket voor onderzoekers over de hele wereld. + Verder verkennen
Het schatten van tumorspecifiek totaal mRNA-niveau voorspelt kankeruitkomsten
 Doping door atleten kan moeilijker te verbergen zijn met nieuwe detectiemethode
Doping door atleten kan moeilijker te verbergen zijn met nieuwe detectiemethode- Ontdekking van periodieke tabellen voor moleculen
- Wat is een dubbele vervangingsreactie?
- Zwavelzuur- en chloorbleekreactie
- Wetenschappers ontwikkelen sondes die zijn ontworpen om de fysieke krachten in levende cellen te onthullen; een wereldprimeur
- Het scheepsverkeer daalde tijdens de eerste maanden van de COVID-pandemie
- Vraag en antwoord:Afvang en opslag van kooldioxide bestuderen tijdens afvalverbranding
- Overstromingen langs de kust van Bay Area veroorzaken regionale verstoringen van woon-werkverkeer
- Rivieren zijn de grootste wereldwijde bron van kwik in oceanen
- Zesvoudige sprong in poolijsverlies verhoogt wereldwijde oceanen
Hoofdlijnen
- Synthetisch muizenembryo met hersenen en kloppend hart gegroeid uit stamcellen
- 30 miljoen jaar oude amfibische beverfossiel is oudste ooit gevonden
- Micro-evolutie: definitie, proces, micro versus macro & voorbeelden
- Waarom is de zon zo helder?
- Wat kan je spit je vertellen over je DNA?
- Wat is een prehistorische toolkit en hoe zou het de menselijke geschiedenis kunnen herschrijven?
- Wat is het doel van de promotor en terminatorregio van de DNA-molecule?
- Wat is de elektrische impuls die een axon naar beneden beweegt?
- Genexpressie in Prokaryotes
- Reconstructie van ijstijddiëten onthult ontrafelend levensweb

- Snellere Salmonella-test verhoogt voedselveiligheid voor mens en dier
- Nauwkeuriger lezen van RNA-modificaties in een zakformaatapparaat

- Videobeelden bieden eerste gedetailleerde observatie van orka's die op witte haaien jagen in Zuid-Afrika

- Microalgen zuiveren water en produceren waardevolle verbindingen


 Deze softwaretitan stelt een computermuseum voor om de rol van Philly bij het starten van de digitale wereld te markeren
Deze softwaretitan stelt een computermuseum voor om de rol van Philly bij het starten van de digitale wereld te markeren- Dynamische beoordeling kan taalleerders helpen om meer succes te hebben
- Onderzoekers ontwikkelen 's werelds kleinste ultrasone detector
- Raciale vooroordelen van blanken kunnen in de loop van de tijd verminderen, uit onderzoek blijkt
- Kosmische uitbarstingen onthullen universums waarin materie ontbreekt
- Hoe een 70 graden hoek te construeren
- Deep-learning AI-systeem zet Singapore op de wereldkaart van big data-analyse
- Studie beoordeelt de mogelijke gevolgen van toekomstige hittegolven voor mensen en dieren in het wild
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
