Wetenschap
Nieuwe techniek zou het modelleren van moleculen veel eenvoudiger kunnen maken
Net als de mensen die ze hebben gemaakt, vinden computers de natuurkunde moeilijk, maar de kwantummechanica nog moeilijker. Maar een nieuwe techniek, ontwikkeld door drie wetenschappers van de Universiteit van Chicago, stelt computers in staat om met veel minder moeite bepaalde uitdagende kwantummechanische effecten in complexe elektronische materialen te simuleren.
Door deze simulaties nauwkeuriger en efficiënter te maken hopen de wetenschappers dat de techniek kan helpen nieuwe moleculen en materialen te ontdekken, zoals nieuwe soorten zonnecellen of kwantumcomputers.
"Deze vooruitgang biedt een enorm potentieel voor het vergroten van ons begrip van moleculaire verschijnselen, met aanzienlijke implicaties voor de scheikunde, materiaalkunde en aanverwante gebieden", zegt wetenschapper Daniel Gibney, Ph.D. van de Universiteit van Chicago. student scheikunde en eerste auteur van het artikel, gepubliceerd op 14 december in Physical Review Letters .
Elektronen en energie
Een blad of een zonnepaneel ziet er van buitenaf glad en eenvoudig uit, maar zoom in op het moleculaire niveau en je ziet een enorm ingewikkelde dans van elektronen en moleculen.
Om nieuwe vooruitgang te boeken op het gebied van duurzaamheid, productie, landbouw en vele andere gebieden, modelleren wetenschappers het gedrag van deze chemische en moleculaire interacties. Dit helpt nieuwe ontwerpmogelijkheden voor de toekomst te onthullen – voor alles, van nieuwe manieren om koolstofdioxide vast te leggen tot nieuwe soorten kwantumbits.
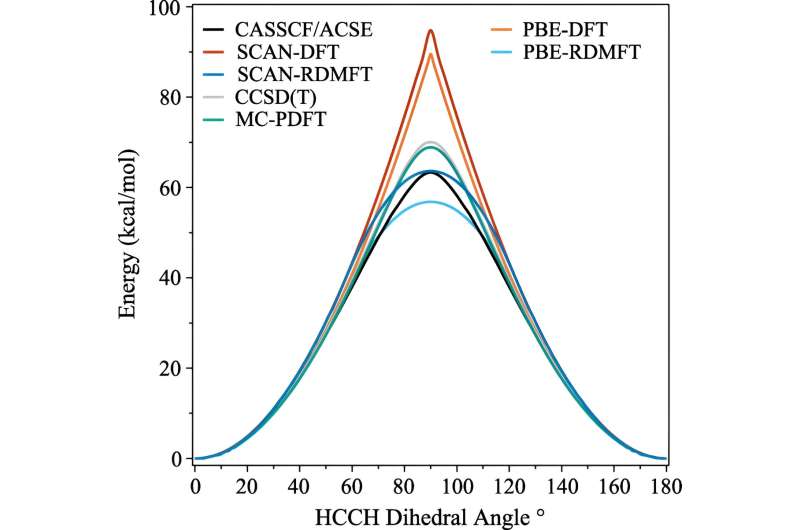
Er zijn de afgelopen decennia veel vooruitgang geboekt, maar een van de gebieden die nog steeds hardnekkig moeilijk te simuleren zijn, is wanneer de moleculen complex kwantummechanisch gedrag gaan vertonen dat wetenschappers sterke correlatie noemen.
Het probleem is dat zodra elektronen beginnen te pronken met hun meest kwantummechanische effecten – zoals ‘verstrengeld raken’ – de berekeningen onmiddellijk veel meer rekenkracht nodig hebben. Zelfs supercomputers hebben moeite met het omgaan met de implicaties.
Een van de meest gebruikte berekeningen is de dichtheidsfunctionaaltheorie. "Dit is feitelijk de meest alomtegenwoordige techniek voor het voorspellen van de elektronische structuur, maar het is in wezen een benadering waarbij alle elektronen worden behandeld als een functie van één elektron", legt David Mazziotti uit, hoogleraar scheikunde en senior auteur van het onderzoek.
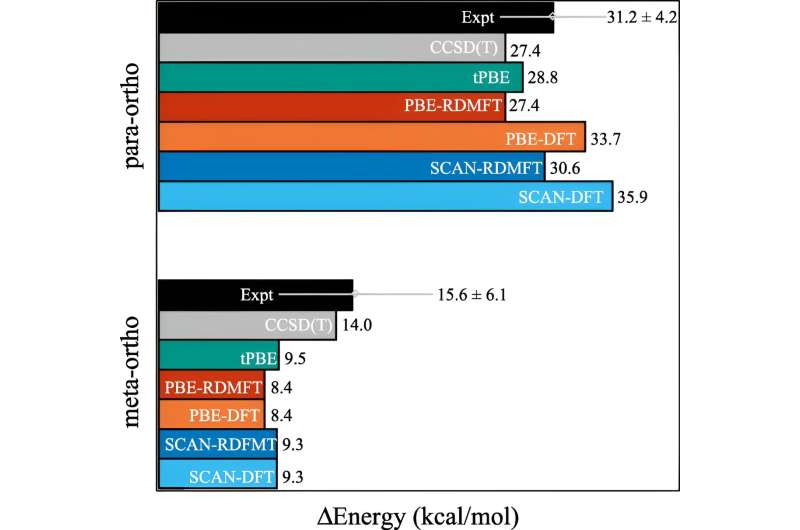
Voor veel berekeningen volstaat een benadering. Maar het begint af te breken naarmate het gedrag van de elektronen meer gecorreleerd raakt, zoals gebeurt wanneer de kwantummechanica een rol gaat spelen. In de kwantummechanica kunnen deze elektronen zich tegelijkertijd op meerdere plaatsen, of orbitalen, bevinden. Dit belemmert niet alleen de menselijke hersenen, maar ook de dichtheidsfunctionaaltheorie.
"En dit is een belangrijk probleem, omdat veel van de problemen waar we in de 21e eeuw om geven – zoals nieuwe moleculen en materialen voor hernieuwbare energie en duurzaamheid – vereisen dat we de kwantumaard van materialen exploiteren", aldus Mazziotti.
Mazziotti, Gibney en derde auteur Jan-Niklas Boyn ontdekten dat ze een universele correctie konden toevoegen aan de dichtheidsfunctionaaltheorie, waardoor de elektronen verstrikt kunnen raken tussen meerdere orbitalen tegelijk.
"Hierdoor kunnen de orbitalen in de berekening niet alleen volledig gevuld of volledig leeg zijn, maar ook ergens daartussenin", aldus Mazziotti. "We komen tot een beeld van één elektron dat nog steeds in staat is het gedrag vast te leggen dat voortkomt uit gecorreleerde elektroneneffecten van meerdere lichamen."
Een 'universele' aanpassing
Als bonus kunnen de wetenschappers zeggen dat de code aan bestaande algoritmen kan worden toegevoegd zonder dat je die code hoeft te herschrijven. "In principe treedt de correctie in werking wanneer dat nodig is, maar interfereert verder niet met de rest van de code", aldus Gibney.
Het is ook universeel:het kan worden toegevoegd aan code die vele soorten elektronisch gedrag simuleert, of het nu gaat om fotovoltaïsche zonnepanelen of koolstofvastlegging of supergeleidende materialen, of zelfs biologie.
Boyn legt bijvoorbeeld uit dat één toepassing zou kunnen liggen in het begrijpen van de chemie die plaatsvindt met behulp van enzymen die metaalatomen bevatten, bekend als metallo-enzymen.
"Er is bijvoorbeeld een overvloed aan metallo-enzymen die verantwoordelijk zijn voor veel van de chemie in je cellen, maar ze zijn notoir moeilijk te beschrijven met de huidige modellen," zei hij. "Deze theorie zou ons in de nabije toekomst in staat kunnen stellen deze chemie aan te pakken op een manier die nu onmogelijk is."
Meer informatie: Daniel Gibney et al, Universele generalisatie van dichtheidsfunctionele theorie voor statische correlatie, Physical Review Letters (2023). DOI:10.1103/PhysRevLett.131.243003
Journaalinformatie: Fysieke beoordelingsbrieven
Aangeboden door Universiteit van Chicago
 bedrog, ontheemde Amerikanen verlangen ernaar naar huis terug te keren na orkaan
bedrog, ontheemde Amerikanen verlangen ernaar naar huis terug te keren na orkaan- In Yakutia, Rusland graaft naar diamanten in permafrost
- 5 leuke Earth Day-spellen voor kinderen
- Hoe Antarctische ijssmelt een omslagpunt kan zijn voor het klimaat van de planeet
- IJslandse vulkaanuitbarsting langste in halve eeuw
Hoofdlijnen
- Moleculaire mechanismen van paaigewoonten voor adaptieve straling van endemische Oost-Aziatische karperachtigen
- Wat is een Punnett-vierkant?
- Waarom blozen mensen?
- Nieuwe aanpak verdubbelt efficiëntie van stamcelbewerking meer dan, melden onderzoekers
- Studie onderzoekt conflict tussen boeren en roofdieren
- In Australië zijn kaketoes en mensen in een wapenwedloop om toegang tot afval
- Mensenhandelaars in wilde dieren en planten zetten hun illegale handel voort tijdens de COVID-lockdown – wat kunnen we leren van hun veerkracht?
- Klimaatverandering zou de verspreiding van kwallen en ander gelatineus zoöplankton in de Noordelijke IJszee aanzienlijk kunnen veranderen
- Rangschikken met behulp van een vierkantswortelcurve
- Onderzoekers ontwikkelen zich snel, elektro-optische modulator ter grootte van een micrometer

- Doorbraak van nanodeeltjes kan ongezien licht opvangen voor conversie van zonne-energie

- Neutronen en een beetje goud onthullen nieuw type kwantumfaseovergang

- Het ontsluiten van de kracht van een moleculespin

- Nieuwe methode maakt gebruik van warmtestroom om verschillende objecten te laten zweven


 Minder verslavende opioïden ontwerpen door middel van chemie
Minder verslavende opioïden ontwerpen door middel van chemie- Een nieuwe benadering voor het verstandig gebruik van kooldioxide uit uitlaatgassen van auto's
- Lift berekenen voor rotorbladen
- Nieuwe studie van planktonschelpen vernietigt decennia-oud begrip van hun vorming en chemie
- Hoe een slingerpsychrometer te lezen
- Ontdekking van geheel nieuwe klasse RNA-caps in bacteriën
- Studies van individuele nanodeeltjes kunnen de sleutel zijn tot toekomstige katalyse
- Een schijnwerper schijnen op de machinerie van het leven
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
