Wetenschap
Machinaal lerende modellen van materie voorbij interatomaire potentialen
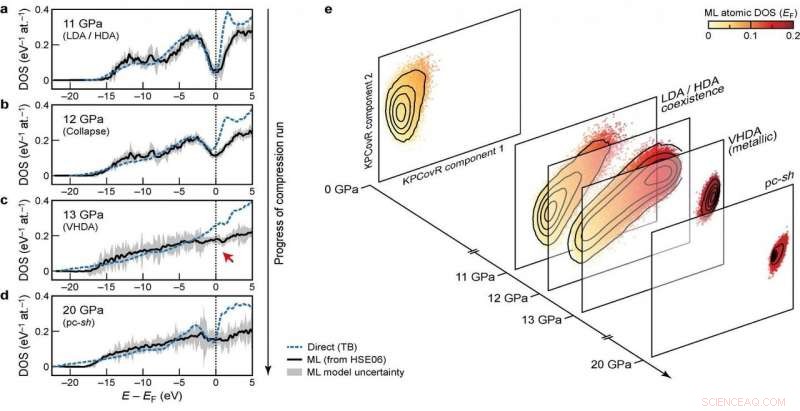
Elektronische densiteiten van staten (DOS) in verschillende stadia van de compressierun Credit:@Michele Ceriotti
Het combineren van elektronische structuurberekeningen en machine learning (ML) technieken is een gangbare benadering geworden in de atomistische modellering van materie. Door de twee technieken samen te gebruiken, konden onderzoekers, bijvoorbeeld, om modellen te maken die atomaire coördinaten gebruiken als de enige invoer om goedkoop elke eigenschap te voorspellen die kan worden berekend door de eerste-principeberekeningen die zijn gebruikt om ze te trainen.
Terwijl de vroegste en inmiddels meest geavanceerde inspanningen gericht waren op het gebruik van voorspellingen van totale energieën en atomaire krachten om interatomaire potentialen te construeren, meer recente inspanningen waren gericht op aanvullende eigenschappen van kristallen en moleculen zoals ionisatie-energieën, NMR chemische afschermingen, diëlektrische responseigenschappen en ladingsdichtheid. In het artikel "Het leren van de elektronische dichtheid van toestanden in gecondenseerde materie, " Ceriotti en collega's richten zich op de elektronische dichtheid van staten (DOS), een andere grootheid die ten grondslag ligt aan veel nuttige materiaaleigenschappen, waarvan sommige experimenteel kunnen worden waargenomen.
De DOS is in wezen het aantal verschillende toestanden dat elektronen kunnen innemen op een bepaald energieniveau, en kan worden gebruikt, bijvoorbeeld, om de elektronische bijdrage aan de warmtecapaciteit in metalen en de dichtheid van vrije ladingsdragers in halfgeleiders te berekenen. Het is een indirecte proxy voor eigenschappen zoals de energiebandkloof, de bandenergie en het optische absorptiespectrum.
"Het voorspellen van de DOS is een interessante oefening op zich, omdat het in wezen de eenvoudigst mogelijke beschrijving is van de elektronische structuur buiten het beeld van de grondtoestand, "Zei Ceriotti. "Het is ook handig omdat er veel eigenschappen zijn die je kunt berekenen vanaf de DOS, waardoor het een goed voorbeeld is van hoe de volgende generatie ML-modellen op een vergelijkbare manier kan worden gebruikt als elektronische structuurberekeningen, ze op een indirecte manier gebruiken om tussenliggende hoeveelheden te berekenen die vervolgens gemakkelijk kunnen worden verwerkt om eigenschappen te evalueren die moeilijker direct te leren zijn."
Bij het ontwikkelen van het model is de groep streefde naar overdraagbaarheid over verschillende fasen en naar schaalbaarheid naar grote systeemgroottes. Hun ultieme aanpak, die kijkt naar hoe verschillende atomaire configuraties de verdeling van energieniveaus beïnvloeden, voldoet aan deze doelen - het was in staat om DFT-berekende DOS te leren en te voorspellen voor een diverse dataset van siliciumstructuren, die een breed scala van thermodynamische omstandigheden en verschillende fasen dekt. Het schaalt ook lineair, in plaats van met de derde macht van het aantal atomen zoals bij elektronische structuurberekeningen, waardoor het toepasbaar is op grote constructies. Eindelijk, het model maakte een analyse van de lokale DOS mogelijk, onderzoekers de kans geven om het samenspel tussen structurele motieven en elektronische structuur te onderzoeken.
De combinatie van overdraagbaarheid, en schaalbaarheid van voorspellingen tot grote systeemgroottes, het model toepasbaar maken om al lang bestaande open vragen in de materiaalkunde aan te pakken. Het nieuwe raamwerk is al gebruikt om de elektronische eigenschappen van een 100.000-atomen simulatie van amorf silicium op te helderen, een reeks faseovergangen ondergaan wanneer gecomprimeerd tot 20 Gpa, in een paper gepubliceerd in Natuur vandaag in samenwerking met een team bestaande uit onderzoekers uit Oxford, Cambridge, het US Naval Research Laboratory en de Ohio University. De voorspelde DOS wordt ook gebruikt om uit te leggen hoe de door druk geïnduceerde structurele transformaties zich koppelen aan de elektronische structuur van het materiaal.
Door het nieuwe model te combineren met een van de gevestigde potentiële energiemodellen, is het ook mogelijk om de elektronische bijdragen aan macroscopische eigenschappen zoals de warmtecapaciteit van metalen te berekenen en om simulaties uit te voeren die rekening houden met eindige elektronische temperatuureffecten, zoals aangetoond in een ander binnenkort te verschijnen artikel waarin de eigenschappen van nikkel bij hoge temperaturen worden besproken. Inderdaad, het nieuwe model is een cruciale stap in de richting van MARVEL's doel om geïntegreerde machine learning-modellen te ontwikkelen die de kostbare elektronische structuurberekeningen aanvullen - en misschien uiteindelijk vervangen.
"Er zijn andere eigenschappen dan de elektronendichtheid van toestanden, zoals optische excitaties, en NMR-reactie, die we met machinaal leren nauwkeurig hebben kunnen voorspellen." structuurberekening, maar tegen een fractie van de kosten."
 Onderzoeksteam pioniert sneller, goedkopere en groenere manier om aminozuren te produceren uit plantaardig afval
Onderzoeksteam pioniert sneller, goedkopere en groenere manier om aminozuren te produceren uit plantaardig afval- Onderzoekers vinden potentieel in milieuvriendelijke synthesizer
- Nieuwe schaal voor elektronegativiteit herschrijft het leerboek scheikunde
- Chemici zetten titanium nanodeeltjes om in een efficiënt wapen tegen vervuiling
- Onderzoekers ontwikkelen microbellenwasser om gevaarlijke biofilms te vernietigen
- NASA-NOAA's Suomi NPP Tropische cycloon Merbok Nnaring Hong Kong
- Het tappen van zoet water onder de oceaan heeft gevolgen
- IJslandse vulkaan zakt na eerste uitbarsting in 900 jaar
- Biotische factoren in het Florida Manatee Ecosystem
- Hoog in de bergen:Andesbossen hebben een groot potentieel om koolstof op te slaan onder klimaatverandering
Hoofdlijnen
- Het gebit van de wedgefish lijkt ontworpen om schaaldieren te pletten, maar hij eet ook pijlstaartroggen
- Lignine-afval aangepast voor industrieel gebruik van bio-olie
- Het unieke pentraxine-koolzuuranhydrase-eiwit reguleert het vermogen van vissen om te zwemmen
- Central Dogma (Gene Expression): Definitie, Stappen, Verordening
- Warme wateren in het noordwesten trekken paaiende vissen naar het noorden
- Hoe Rigor Mortis te onderscheiden van een Cadaveric Spasm
- Pittige Tomaten,
- Wat zijn de vier eukaryotische koninkrijken?
- Virusstamping - een veelzijdige nieuwe methode voor genetische manipulatie van afzonderlijke cellen




 Latijn kan studenten helpen hun moedertaal te overbruggen met Engels
Latijn kan studenten helpen hun moedertaal te overbruggen met Engels- Microplastische verontreiniging gevonden in gemeenschappelijke bron van grondwater, onderzoekers rapporteren
- Race om dieren te redden op de door brand verwoeste Galapagos in Australië
- Deze oplossing voor economische theorie verandert alles, van gokken tot Ponzi-schema's
- Het magnetisme van een grafeen driehoekige vlok ontrafelen
- Het pad verlichten voor beeldvorming met superresolutie met verbeterde rhodamine-kleurstoffen
- Astronomen meten 3D-stellaire beweging met recordprecisie (update)
- De rol van schedelmodificatie bij identiteitsvorming
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
