Wetenschap
Onderzoekers werpen neurale netten om moleculaire beweging te simuleren
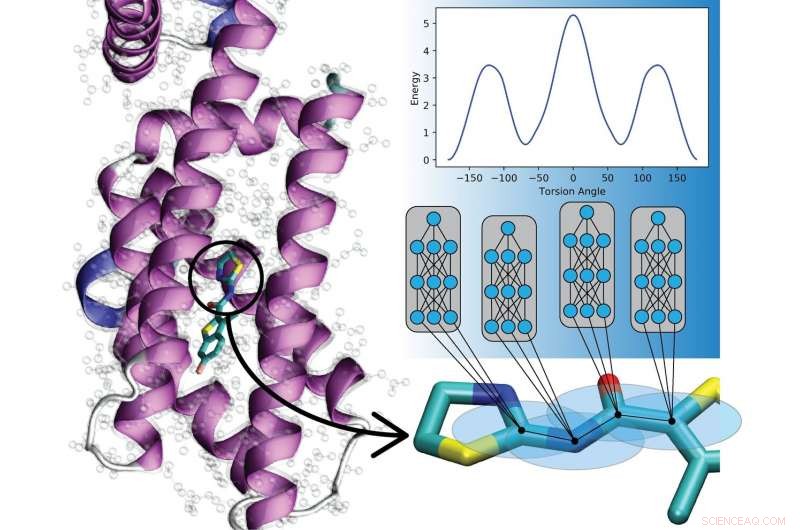
Nieuwe deep learning-modellen voorspellen de interacties tussen atomen in organische moleculen. Deze modellen zullen computationele biologen en onderzoekers van geneesmiddelenontwikkeling helpen ziekten te begrijpen en te behandelen. Krediet:Los Alamos Nationaal Laboratorium
Nieuw werk van Los Alamos National Laboratory, de Universiteit van North Carolina in Chapel Hill, en de Universiteit van Florida laat zien dat kunstmatige neurale netten kunnen worden getraind om kwantummechanische wetten te coderen om de bewegingen van moleculen te beschrijven, superchargen-simulaties mogelijk over een breed scala van velden.
"Dit betekent dat we materialen en moleculaire dynamica nu miljarden keren sneller kunnen modelleren in vergelijking met conventionele kwantummethoden, met behoud van hetzelfde nauwkeurigheidsniveau, " zei Justin Smit, Los Alamos natuurkundige en Metropolis Fellow in de theoretische afdeling van het laboratorium. Begrijpen hoe moleculen bewegen is van cruciaal belang om hun potentiële waarde voor de ontwikkeling van geneesmiddelen te benutten, eiwitsimulaties en reactieve chemie, bijvoorbeeld, en zowel kwantummechanica als experimentele (empirische) methoden worden meegenomen in de simulaties.
De nieuwe techniek, het ANI-1ccx-potentieel genoemd, belooft de mogelijkheden van onderzoekers op veel gebieden te vergroten en de nauwkeurigheid van op machine learning gebaseerde mogelijkheden te verbeteren in toekomstige studies van metaallegeringen en detonatiefysica.
Kwantummechanische (QM) algoritmen, gebruikt op klassieke computers, kan nauwkeurig de mechanische bewegingen van een verbinding in zijn operationele omgeving beschrijven. Maar QM schaalt erg slecht met verschillende molecuulgroottes, waardoor de reikwijdte van mogelijke simulaties ernstig wordt beperkt. Zelfs een kleine toename van de molecuulgrootte binnen een simulatie kan de rekenlast dramatisch verhogen. Dus beoefenaars nemen vaak hun toevlucht tot empirische informatie, die de beweging van atomen beschrijft in termen van klassieke fysica en de wetten van Newton, simulaties mogelijk maken die opschalen naar miljarden atomen of miljoenen chemische verbindingen.
traditioneel, empirische mogelijkheden hebben een afweging moeten maken tussen nauwkeurigheid en overdraagbaarheid. Wanneer de vele parameters van de potentiaal nauwkeurig zijn afgestemd op één verbinding, de nauwkeurigheid neemt af op andere verbindingen.
In plaats daarvan, het Los Alamos-team, met de Universiteit van North Carolina in Chapel Hill en de Universiteit van Florida, heeft een machine learning-benadering ontwikkeld, transfer learning genaamd, waarmee ze empirische mogelijkheden kunnen opbouwen door te leren van gegevens die zijn verzameld over miljoenen andere verbindingen. De nieuwe benadering met het empirische potentieel van machine learning kan in milliseconden worden toegepast op nieuwe moleculen, onderzoek naar een veel groter aantal verbindingen mogelijk maken over veel langere tijdschalen.
 Licht werpen op het reactiemechanisme van PUVA-lichttherapie voor huidziekten
Licht werpen op het reactiemechanisme van PUVA-lichttherapie voor huidziekten- Instant waterreinigingsmethode miljoenen keren beter dan commerciële aanpak
- Onderzoekers ontwikkelen een efficiënte, energiezuinige methode voor het upcyclen van polyethyleen plastic afval tot waardevolle moleculen
- Waarom uien ons aan het huilen maken (en waarom sommigen niet)
- Cryo-elektronenmicroscopie bereikt ongekende resolutie met behulp van nieuwe rekenmethoden
- Tropische storm, rivier overstroming hamer Golf milieu
- Plastic afval valt uiteen in nanodeeltjes, studie vondsten
- Nieuwe studie onthult hogere microplastics in de lucht van Londen in vergelijking met andere steden
- De keerzijde van de Bitcoin:hoe blockchain duurzame energie kan ondersteunen
- NASA ziet Ewiniar terugglijden in de Zuid-Chinese Zee en sterker worden
Hoofdlijnen
- Pompoengenomen gesequenced, ongewone evolutionaire geschiedenis onthullen
- Wat is perifeer bloed?
- Babyneushoorn galoppeert naar publiek in Singapore Zoo
- Onverwachte regulatie van transcriptiefactoren die cruciaal zijn voor ontwikkeling
- Selenium zou de sleutel kunnen zijn tot het mysterie van kribbebijten bij paarden
- Eukaryotische cel: definitie, structuur en functie (met analogie en diagram)
- Wat zijn twee voorbeelden van reacties Organismen weergeven om de homeostase te onderhouden?
- Wat is een Mordant in de microbiologie?
- Hoe de Amoeben zich reproduceren?
- Nieuwe methode gebruikt ruis om spectrometers nauwkeuriger te maken

- Gebruik van volume in het dagelijks leven

- Tijdkristallen - hoe wetenschappers een nieuwe staat van materie creëerden

- Centripetal Force

- Onderzoekers ontdekken de fysieke oorsprong van elektronische fasescheidingsverschijnselen in complexe oxiden

 Grote nieuwe studie toont oude internationale handelsroutes tussen Exeter en Europa
Grote nieuwe studie toont oude internationale handelsroutes tussen Exeter en Europa- Nieuwe tool ondersteunt toekomst van biologische landbouw
- Gedetailleerde kijk op vroegste momenten van supernova-explosie
- Spacewalkers voltooien zonnepaneelvoorbereiding voor powerboost van station
- Mexico verhoogt alarmniveau als vulkaan as spuwt lava
- Nieuw onderzoek kan leiden tot efficiëntere opslag van elektrische energie
- Amazon Prime Day wordt een fenomeen nu rivalen binnenspringen
- Bionische katalysatoren om schone energie te produceren
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
