Wetenschap
Doorbraak gemeld in door machine learning verbeterde kwantumchemie
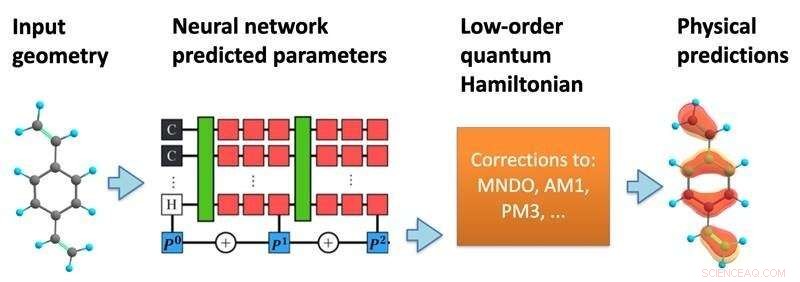
De structuur van het model. Een neuraal netwerk verwerkt een moleculaire geometrie om een semi-empirische kwantum Hamiltoniaan te voorspellen, die vervolgens zelfconsistent wordt opgelost om een verscheidenheid aan chemische eigenschappen te voorspellen. Krediet:Kipton Barros, Los Alamos National Laboratory.
In een nieuwe studie, gepubliceerd in Proceedings of the National Academy of Sciences , hebben onderzoekers van het Los Alamos National Laboratory voorgesteld om meer wiskunde van de kwantummechanica op te nemen in de structuur van de voorspellingen van machine learning. Met behulp van de specifieke posities van atomen in een molecuul, voorspelt het machine learning-model een effectieve Hamilton-matrix, die de verschillende mogelijke elektronische toestanden beschrijft samen met de bijbehorende energieën.
Vergeleken met traditionele simulaties van kwantumchemie, maakt de op machine learning gebaseerde benadering voorspellingen tegen veel lagere rekenkosten. Het maakt kwantitatief nauwkeurige voorspellingen mogelijk met betrekking tot materiaaleigenschappen, geeft een interpreteerbaar inzicht in de aard van chemische bindingen tussen atomen en kan worden gebruikt om andere complexe fenomenen te voorspellen, zoals hoe het systeem zal reageren op verstoringen, zoals interacties tussen licht en materie. De methode biedt ook een sterk verbeterde nauwkeurigheid ten opzichte van traditionele machine learning-modellen, en toont succes in overdraagbaarheid, d.w.z. het vermogen van het model om voorspellingen te doen die veel verder gaan dan de gegevens die de basis vormden van de training.
De vergelijkingen van de kwantummechanica bieden een routekaart voor het voorspellen van de eigenschappen van chemicaliën, uitgaande van fundamentele wetenschappelijke theorieën. Deze vergelijkingen kunnen echter snel te duur worden in termen van computertijd en -kracht wanneer ze worden gebruikt om gedrag in grote systemen te voorspellen. Machine learning biedt een veelbelovende aanpak om dergelijke grootschalige simulaties te versnellen. Het gebruik van machinaal leren om chemische eigenschappen te voorspellen, biedt het potentieel voor grote technologische vooruitgang, met toepassingen van schonere energie tot sneller ontwerp van farmaceutische geneesmiddelen. Dit is een zeer actief onderzoeksgebied, maar de meeste bestaande benaderingen gebruiken eenvoudige en heuristische benaderingen voor het ontwerp van de machine learning-modellen.
In hun onderzoek hebben de onderzoekers aangetoond dat machine learning-modellen de basisstructuur van de fundamentele natuurwetten kunnen nabootsen. Deze wetten kunnen erg moeilijk direct te simuleren zijn. De machine learning-benadering maakt voorspellingen mogelijk die gemakkelijk te berekenen zijn en nauwkeurig zijn in een breed scala aan chemische systemen.
Het verbeterde machine learning-model kan snel en nauwkeurig een breed scala aan eigenschappen van moleculen voorspellen. Deze benaderingen scoren zeer goed op belangrijke benchmarks in computationele chemie en laten zien hoe deep learning-methoden kunnen blijven verbeteren door meer gegevens uit experimenten op te nemen. Het model kan ook slagen in uitdagende taken zoals het voorspellen van de dynamiek van de aangeslagen toestand - hoe systemen zich gedragen met verhoogde energieniveaus. Deze tool is een baanbrekende mogelijkheid voor de kwantumchemie. Het zal onderzoekers in staat stellen de reactiviteit en opgewonden toestanden van nieuwe moleculen beter te begrijpen. + Verder verkennen
Computers blinken uit in scheikunde
 Nauwkeurige en efficiënte dynamische rekenstrategie voor heterogene katalyse
Nauwkeurige en efficiënte dynamische rekenstrategie voor heterogene katalyse- Chemici tonen aan dat natrium veilig kan worden gebruikt voor kruiskoppelingsreacties
- Meelwormen koken tot een smakelijke, gezonde, vleesachtige smaakmaker
- Geminiaturiseerde neuroprobe voor het nemen van monsters van neurotransmitters in de hersenen
- COVID-19:een wake-up call om de toeleveringsketen van geneesmiddelen opnieuw in evenwicht te brengen?
- Studie identificeert nieuwe route voor smeltwater van Groenland om de oceaan te bereiken
- Steden, stammen proberen een nieuwe milieuaanpak:natuurrechten geven
- Tsunamiwacht voor Hawaï opgeheven na aardbeving in het noorden van de Stille Oceaan
- Grote stevige producten bovenaan lijst met slechtste plastic afval:rapport
- Onderzoek wordt realiteit in onderzoek naar de impact van brand op de waterbronnen van sonoma
Hoofdlijnen
- Hoe beïnvloedt stress je hersens?
- Onderzoek toont aan dat bodemradar fijne wortels in gewassen kan detecteren
- Nieuwe Dehalogenimonas sp. stam kan dechlorering van diclofenac stimuleren
- Een sleutel vinden om geblokkeerde differentiatie in microRNA-deficiënte embryonale stamcellen te ontgrendelen
- Huidige vee-injecties verhogen het risico op letsel, onderzoek vindt
- Baanbrekende ontdekking van een geurdetecterende receptorversterker
- Wat zijn 3 functies van de navelstreng?
- Vrouwelijke mangoesten helpen hun pups door rivalen te verdrijven
- Hoe werken vaccins met het immuunsysteem?






 2017 Noord-Koreaanse kernproef 10 keer groter dan eerdere tests, nieuwe studie vondsten
2017 Noord-Koreaanse kernproef 10 keer groter dan eerdere tests, nieuwe studie vondsten- Zilveren nanodeeltjes kunnen ooit de sleutel zijn tot apparaten die harten sterk en stabiel laten kloppen
- Hoe overleven reuzenpanda's?
- Bibliotheken kunnen 3D-printers hebben, maar ze gaan nog steeds over boeken
- Beschrijving van de verschillende soorten wolken
- Risicovolle strategie voor de planeet kunstmatig koelen, nieuw onderzoek toont aan
- AkzoNobel spettert uit terwijl de Q1-winst een rooskleurig beeld schetst
- een nieuwe, natuurlijke wascoating maakt kleding waterafstotend en ademend
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
