Wetenschap
Van chemische grafieken tot structuren
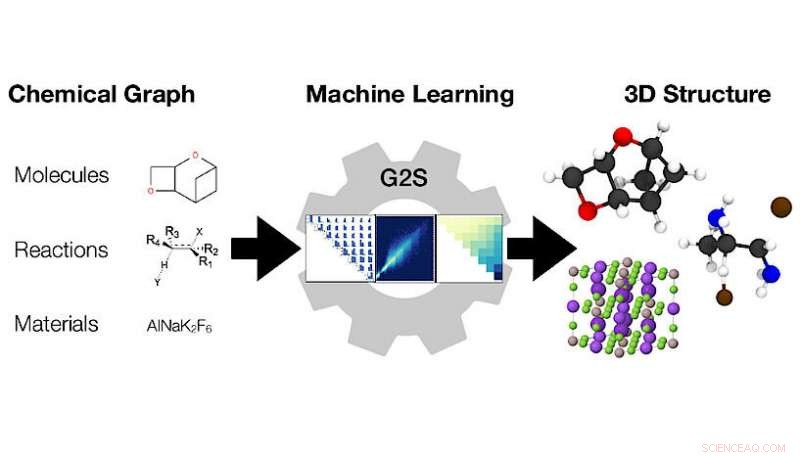
Het machine learning-model Graph2Structure gebruikt grafieken van chemische verbindingen (links) om hun 3D-coördinaten te voorspellen (rechts). Krediet:Dominik Lemm, Universiteit van Wenen
3D-configuraties van atomen bepalen alle materiaaleigenschappen. Kwantitatieve voorspellingen van nauwkeurige evenwichtsstructuren, 3D-coördinaten van alle atomen, van een chemische grafiek, een weergave van de structuurformule, is een uitdagende en rekenkundig dure taak die aan het begin staat van vrijwel elke computationele chemie-workflow. Onderzoekers van de Universiteit van Wenen hebben nu een nieuw op machine learning gebaseerd model ontwikkeld om dure berekeningen te verkorten om structuren direct uit grafieken te voorspellen. De nieuwe methode voor "Machine learning-gebaseerde energievrije structuurvoorspellingen van moleculen, overgangstoestanden, en vaste stoffen" wordt gepresenteerd in het laatste nummer van Natuurcommunicatie .
Hoewel vaak afgeschilderd als rigide, chemische verbindingen zijn flexibele driedimensionale objecten die bestaan uit atomen die continu bewegen en oscilleren. Cyrus Levinthal merkte al in 1969 op dat de grote hoeveelheid vrijheidsgraden van chemische verbindingen formeel leidt tot een catastrofaal groot aantal mogelijke conformaties, tot wel 10, 300 (Levinthals Paradoxon). Binnen experimentele waarnemingen, echter, 3D-configuraties van atomen komen overeen met goed gedefinieerde minima van vrije energie en dicteren daardoor alle materiaaleigenschappen. Het paradigma dat structuur de functie bepaalt, is de sleutel voor het bepalen van geneesmiddelinteracties, het optimaliseren van katalysatoren of reacties, en materialen ontdekken. Als gevolg hiervan, in de meeste computationele high-throughput screeningcampagnes (een methode voor snelle wetenschappelijke experimenten), alleen de meest stabiele configuraties zijn gewild. Afhankelijk van het niveau van verfijning binnen de benaderingen die zijn gemaakt bij het schatten van de stabiliteit van materialen, rekenkosten kunnen variëren van minuten tot uren of zelfs dagen voor de berekening van een enkele structuur. Gezien de uitgestrektheid van de ruimte van chemische verbindingen, de ruimte bevolkt door alle denkbare verbindingen (geschat op meer dan 1, 060) vormt deze afweging tussen kosten en kwaliteit een groot knelpunt in het veld.
Onderzoekers van de Universiteit van Wenen onder leiding van Anatole von Lilienfeld pakten dit probleem vanuit een ander perspectief aan, het ontwikkelen van een nieuwe methode die gebruikmaakt van gegevens en universeel toepasbaar is op elke vorm van chemie. Hun nieuwe methode, Grafiek2Structuur, gebruikt kwantumchemische gegevens van hoge kwaliteit om machinale leermodellen te trainen die nieuwe 3D-structuren kunnen voorspellen voor moleculaire grafieken van onzichtbare verbindingen. Deze directe toewijzing van een moleculaire grafiek aan een specifieke 3D-configuratie stelt het model in staat om elke vorm van energieminimalisatie effectief te omzeilen, wat leidt tot een versnelling van meer dan een miljoen in vergelijking met de conventionele methoden. "De mogelijkheid om structuren van hoge kwaliteit te genereren, versnelt niet alleen moleculair ontwerp met hoge doorvoer, maar versnelt ook de dagelijkse workflow, " zegt hoofdauteur van de studie in Natuurcommunicatie Dominik Lemm. "Betrouwbaar genereren van 3D-structuren voor zelfs exotische chemie, zoals open-shell systemen of overgangstoestanden, is een van de moeilijkste taken in atomistische simulatie."
Verdere bevindingen suggereren dat de gegenereerde structuren direct kunnen worden gebruikt als input voor de daaropvolgende evaluatie van op machine learning gebaseerde voorspellingsmodellen voor eigenschappen, waardoor een moleculaire grafiek op een rigoureuze en effectievere manier wordt gekoppeld aan een structuurafhankelijke eigenschap.
 Glas verwerken als een polymeer
Glas verwerken als een polymeer- Nieuw geavanceerd materiaal vertoont buitengewone stabiliteit over een breed temperatuurbereik
- Hoe PH van bufferoplossingen te berekenen
- Opwekking van waterstof op zonne-energie efficiënter maken in microzwaartekracht
- Hoe fractionele overvloed van een isotoop te vinden
- Drone- en landsat-beelden tonen langdurige verandering in vegetatiebedekking langs intermitterende rivier
- Sommige bomen kunnen de zeespiegelstijging niet overleven
- Onderzoekers vinden verband tussen Atlantische orkanen en weersysteem in Oost-Azië
- Koeien milieuvriendelijker maken
- EPA's COVID-19-beleid zal de toegang van het publiek tot klimaatgegevens verder beperken
Hoofdlijnen
- 7 soorten bindweefsel
- EU stemt ermee in om in 2018 meer vis op een duurzame manier te vangen
- Hoe SARS werkt
- Wat gebeurt er als er sprake is van mitose?
- Ontdekking helpt de nauwkeurigheid van CRISPR-Cas9-genbewerking te verbeteren
- Zijn gelukkige mensen gezonder?
- NASA Twins-onderzoek wordt gerepliceerd op Everest
- Wat doet het endoplasmatisch reticulum?
- Carnivoren weten dat het eten van andere karkassen van carnivoren ziekten overdraagt
- Na 90 jaar, wetenschappers onthullen de structuur van benzeen
- U uit Minnesota krijgt subsidie om beter plastic op basis van maïs te ontwikkelen

- Meer van methaan maken
- 5 manieren om te weten of er een chemische verandering heeft plaatsgevonden

- Als je beter kijkt, zie je axiale filamenten in spicules van zeespons die uit eiwitten bestaan

 Nanodeeltjes geproduceerd door het verbranden van steenkool leiden tot schade aan de longen van muizen, suggereert toxiciteit voor mensen
Nanodeeltjes geproduceerd door het verbranden van steenkool leiden tot schade aan de longen van muizen, suggereert toxiciteit voor mensen- Waarom de revolutie van elektrische voertuigen zijn eigen problemen met zich mee zal brengen
- Hoe een defecte solenoïde te detecteren
- 3D virtueel snijden van een antieke viool onthult oude lakmethoden
- Mansplaining:nieuwe oplossingen voor een vervelend oud probleem
- Calculus III voor cellen
- Laag opleidingsniveau het duidelijkste kenmerk van wanbetalingen
- Waarom heeft het universum zichzelf niet vernietigd? Neutrino's kunnen het antwoord bevatten
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- Italian | Portuguese | Swedish | German | Dutch | Danish | Norway | French | Spanish |

-
Wetenschap © https://nl.scienceaq.com
