Wetenschap
Röntgenabsorptiespectra voorspellen uit grafieken
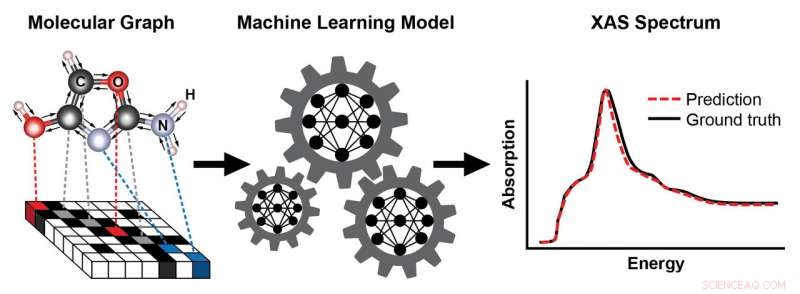
Een schema dat de stappen toont voor het trainen van een machine learning-model om een röntgenabsorptiespectrum (XAS) te voorspellen op basis van de bekende structuur van een molecuul. De structuur van het molecuul wordt weergegeven als een grafiek, met atomen als knopen en chemische bindingen als randen. Deze weergave legt de connectiviteit van atomen vast - hier, koolstof (C), zuurstof (O), stikstof (N), en waterstof (H) - en het type en de lengte van de chemische bindingen die ze verbinden. Het resulterende XAS-spectrum bevat rijke informatie over de lokale chemische omgeving van absorberende atomen, zoals hun symmetrie en het aantal naburige atomen. Krediet:Brookhaven National Laboratory
Röntgenabsorptiespectroscopie (XAS) is een populaire karakteriseringstechniek voor het onderzoeken van de lokale atomaire structuur en elektronische eigenschappen van materialen en moleculen. Omdat atomen van elk element röntgenstralen absorberen met karakteristieke energieën, XAS is zeer geschikt om de ruimtelijke verdeling van elementen in een steekproef in kaart te brengen. Typisch, wetenschappers voeren XAS-experimenten uit bij synchrotron-lichtbronnen - zoals de National Synchrotron Light Source II (NSLS-II) - omdat ze zeer heldere, afstembare röntgenstralen. Door de absorptie in een monster te meten bij verschillende röntgenstralingsenergieën, wetenschappers kunnen een plot genereren dat een röntgenabsorptiespectrum wordt genoemd.
"XAS is een belangrijke mogelijkheid voor gebruikers van het Brookhaven National Laboratory's NSLS-II en het Center for Functional Nanomaterials (CFN), beide U.S. Department of Energy (DOE) Office of Science User Facilities die openstaan voor de wetenschappelijke onderzoeksgemeenschap, " zei Deyu Lu, een natuurkundige in de CFN Theory and Computation Group. "Met de juiste analysetools XAS kan enorme inzichten bieden in nanowetenschappelijk onderzoek. De ontwikkeling van dergelijke tools staat centraal in onze missie als gebruikersfaciliteiten."
Classificatie van lokale chemische omgevingen
Verschillende gebieden van het röntgenabsorptiespectrum zijn gevoelig voor verschillende aspecten van de materiaaleigenschappen in een monster. Bijvoorbeeld, de röntgenabsorptie-near-edge-structuur (XANES) richt zich op het nabije-randgebied van het spectrum, net boven de aanvangsenergie die voldoende is om een elektron uit de binnenste schillen van een atoom te exciteren naar een lege toestand. XANES codeert voor rijke informatie over de lokale chemische omgeving van absorberende atomen in een monster, inclusief hun geometrische coördinatie, symmetrie, en laadtoestand (het aantal elektronen dat wordt gewonnen of verloren door chemische binding). Maar het analyseren van spectrale gegevens is zeer uitdagend vanwege hun abstracte aard.
"In tegenstelling tot een microscoopopname van een materiaal waar je direct kenmerken zoals kristalliniteit of defecten kunt zien, XANES-spectra coderen informatie die domeinexpertise vereist om te interpreteren, " legde Lu uit.
Standaardinterpretatie van signalen in een XANES-spectrum is gebaseerd op karakteristieke kenmerken die bekend staan als "vingerafdrukken, " die zijn opgebouwd uit metingen op referentiematerialen. Echter, deze vingerafdrukbenadering mislukt wanneer het monster geen eenvoudig kristal is en relevante referentiematerialen niet gemakkelijk kunnen worden geïdentificeerd.
Grootschalige, op theorie gebaseerde simulaties van atomaire structuurmodellen kunnen zeer bruikbare inzichten opleveren voor de interpretatie van experimentele XANES-spectra; echter, deze simulaties zijn vaak rekenkundig duur en tijdrovend, en hun nauwkeurigheidsniveau hangt sterk af van de gekozen theoretische benaderingen en het bestudeerde systeem. Als resultaat, robuuste spectrale interpretatie is momenteel het knelpunt van XAS-onderzoeken. Verder, real-time interpretatie van XAS-spectra is naar voren gekomen als een nieuwe uitdaging voor studies van de dynamische evolutie van materialen onder bedrijfsomstandigheden en autonoom experimenteren. De behoefte aan robuuste, efficiënte spectrale interpretatie wordt steeds wijdverbreid bij synchrotron-lichtbronnen.
"Echte tijd, nauwkeurige interpretatie van röntgenverstrooiing en spectroscopiemetingen zoals röntgenabsorptie, fluorescentie, en diffractie is een belangrijk vermogen voor gebruikers die onderzoek doen bij NSLS-II en andere synchrotron-lichtfaciliteiten, " zei Mehmet Topsakal, een wetenschappelijk medewerker in de Materials for Energy Applications Group van Brookhaven's Nuclear Science and Technology Department, die geavanceerde data-analyse en machine learning-technieken voor röntgenspectroscopie ontwikkelt. "Elk jaar, duizenden wetenschappers van over de hele wereld komen naar NSLS-II om de eigenschappen van verschillende materialen te onderzoeken. Een ultramoderne pijplijn voor spectrale analyse zou gebruikers in staat stellen om nuttige feedback over hun monsters te verkrijgen terwijl experimenten aan de gang zijn en om on-the-fly aanpassingen te maken om experimenten te begeleiden. De vraag is, hoe kunnen we spectrale interpretatie in realtime doen om structuur-spectrumcorrelaties te ontdekken?"
Informatie extraheren met machine learning
Gebruikmakend van big data en machine learning, Lu en Topsakal probeerden deze vraag te beantwoorden met computerwetenschapper Shinjae Yoo van Brookhaven Lab's Computational Science Initiative (CSI) en Columbia University Ph.D. kandidaat en DOE Computational Science Graduate Fellow Matthew Carbone.
"De DOE Computational Science Graduate Fellowship heeft me een unieke kans geboden om verder te gaan dan mijn Ph.D.-onderzoek in de chemische fysica aan Columbia om de kracht van machine learning-algoritmen te onderzoeken, werken samen met Brookhaven-wetenschappers, "zei Carbone. "Machine learning maakt gebruik van enorme datasets om zeer scherpzinnige modellen te bouwen die, eenmaal getraind, kan on-the-fly voorspellingen doen op nieuwe gegevens. Dergelijke modellen kunnen worden gebruikt om dure kwantumchemische berekeningen te omzeilen en ondersteuning te bieden bij de karakterisering van operandomateriaal."
Leden van dit team en medewerkers hebben jarenlang gewerkt aan het in kaart brengen van spectrum naar structuur en structuur naar spectrum. in 2017, ze ontwikkelden machine learning-modellen om de gemiddelde coördinatiegetallen van metalen nanodeeltjes uit XANES-spectra te voorspellen. Vorig jaar, ze creëerden een XANES-database om de lokale structuur van een amorfe titaniumoxidecoating voor fotokatalytische toepassingen op te lossen. Ze bouwden ook een machine learning-model dat in staat is om de lokale symmetrie van absorberende atomen te voorspellen uit gesimuleerde XANES-spectra van overgangsmetaaloxiden.
"Bij het uitvoeren van spectrale interpretatie op basis van domeinexpertise, we hebben de neiging om ons te concentreren op specifieke functies die zijn ontwikkeld vanuit onze intuïtie, "Zei Lu. "Machineleren kan de informatie die we nodig hebben op een statistisch opvallende manier extraheren die menselijke vooroordelen elimineert."
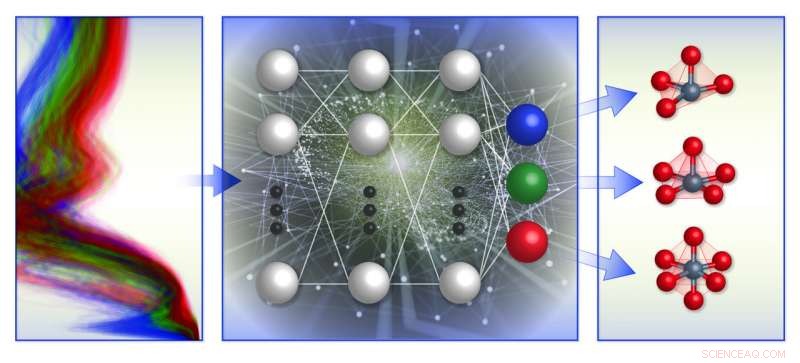
Een schematische illustratie van het op spectrum gebaseerde classificatiekader voor lokale chemische omgevingen van het team. Ze trainden machine learning-modellen (midden) met computationele röntgenabsorptiespectra-database (links) om de lokale geometrie rond positief geladen overgangsmetaalionen (rechts) te voorspellen. Krediet:Brookhaven National Laboratory
Het voorspellen van röntgenabsorptiespectra
Voortbouwend op hun eerdere successen, het team nam een uitdagender probleem aan:een machine learning-model trainen om snel spectra te voorspellen op basis van bekende moleculaire structuren. Een dergelijk model zou de noodzaak van rekenkundig dure simulaties omzeilen, die niet haalbaar zijn tijdens operando-experimenten, wanneer wetenschappers materialen bestuderen onder bedrijfsomstandigheden. Ondanks toenemende inspanningen op het gebied van machine learning om de chemische eigenschappen van materialen te voorspellen, directe voorspellingen van de spectrale functies van echte materialen waren nog niet bereikt.
"Een technische moeilijkheid is het bouwen van een optimale weergave van moleculaire structuren die de inherente symmetrie van de moleculen kunnen coderen als invoerkenmerken voor het machine learning-model, "zei Yo.
Door een recent idee aan te nemen dat is voorgesteld door wetenschappers van Google, Topsakal en Carbone bouwden een machine learning-model op basis van een grafische weergave van moleculen als invoer, waarbij atomen worden weergegeven als knopen en chemische bindingen als randen.
"Computers kunnen moleculen niet zien zoals wij, " zei Topsakal. "Een grafiek is een natuurlijke manier om de structuur en connectiviteit van een molecuul te coderen - door vast te leggen welke atomen zijn verbonden en het type en de lengte van de chemische bindingen die ze verbinden. Bovendien, deze representatie is invariant voor transformaties zoals translaties en rotaties. Dit concept is analoog aan dat in beeldherkenning, waar een object zoals een kat of hond op een achtergrond nog steeds correct kan worden geclassificeerd nadat het beeld is getransformeerd."
Om het model te trainen voor een proof-of-principle demonstratie, het team gebruikte een gevestigde database (genaamd QM9) met berekende structurele en chemische informatie over 134, 000 kleine moleculen met maximaal negen zware atomen per atoomtype (koolstof, stikstof, zuurstof, en fluor). Uit deze databank, ze selecteerden twee trainingssubsets:een subset met moleculen die ten minste één zuurstofatoom bevatten, en een andere subset met moleculen die ten minste één stikstofatoom bevatten - en hun overeenkomstige XANES-spectra berekend. Vervolgens, ze gebruikten hun getrainde modellen om de XANES-spectra te voorspellen voor zuurstof- en stikstofabsorptieranden die overeenkomen met excitaties van elektronen in de binnenste schil van de respectieve atomen.
Het machine learning-model reproduceerde bijna alle significante absorptiepieken en voorspelde de piekposities (energieën waarop pieken verschijnen) en hoogten (absorptie-intensiteiten) met zeer hoge nauwkeurigheid. Het model pikte ook automatisch de domeinkennis op dat röntgenabsorptiespectroscopie gevoelig is voor functionele groepen, of groepen atomen met vergelijkbare chemische eigenschappen en reactiviteit. Afhankelijk van tot welke functionele groep het absorberatoom behoort, verschillende functies verschijnen in de spectra.
"We zijn de eersten die aantonen dat een machine learning-model kan worden gebruikt om de volledige spectrale functies van echte fysieke systemen rechtstreeks vanuit hun structuren nauwkeurig te voorspellen. " zei Topsakal. "Hoewel we ons in onze studie concentreerden op röntgenabsorptiespectroscopie, deze methode kan worden gegeneraliseerd om spectrale informatie voor andere populaire technieken te voorspellen, inclusief infrarood- en gammastralingsspectroscopie."
"Zodra we het machine learning-model hebben getraind, we hoeven geen tijdrovende fysieke simulaties uit te voeren, die minuten duren, uur, of zelfs dagen, " zei Yoo. "We hebben niet alleen realtime spectra-voorspelling mogelijk gemaakt, maar ook de gelijktijdige generatie van honderden en duizenden spectra-inferenties door gebruik te maken van meerdere grafische verwerkingseenheden, of GPU's. Dergelijke technologie is essentieel om geautomatiseerde bundellijncontroles mogelijk te maken en wetenschappelijke ontdekkingen te versnellen. Gecombineerd met methoden om materiaalstructuren te bemonsteren, dergelijke modellen kunnen worden gebruikt om snel relevante structuren te screenen om materiaalontwerp en ontdekking te stimuleren."
Volgende, het team wil concepten combineren uit hun model dat lokale symmetrie voorspelt uit XANES-spectra en dit nieuwe model dat XANES-spectra voorspelt uit moleculaire structuren. uiteindelijk, hun doel is om meer uitgebreide informatie over de lokale chemische omgeving of zelfs de structuur van hele moleculen te extraheren uit experimentele metingen.
"Machine leermiddelen, zoals die voor beeld- en spraakherkenning en het ontdekken van geneesmiddelen, zijn volop in ontwikkeling, " zei Lu. "De sleutel is om uit te zoeken hoe deze tools op een innovatieve manier kunnen worden aangepast om materiaalwetenschappelijke problemen aan te pakken."
"Ons doel bij het ontwikkelen van kunstmatige intelligentie en machine learning-technologieën is om unieke wetenschappelijke uitdagingen op te lossen door zowel de nieuwste technologische doorbraken op deze gebieden toe te passen als nieuwe benaderingen te bedenken die bijdragen aan de respectieve onderzoeksgemeenschappen, " voegde Yo toe.
 Het werk aan een zeldzame molecule heeft tot doel de celtherapie te verbeteren en een functionele genezing voor HIV te leveren
Het werk aan een zeldzame molecule heeft tot doel de celtherapie te verbeteren en een functionele genezing voor HIV te leveren- Wat is de CO2-voetafdruk van een plastic fles?
- Immunotherapie voor dodelijke bacteriën toont vroege belofte
- Kristaloorlogen:onderzoek kan leiden tot efficiëntere methoden voor kristaltechnologie
- Nieuwe tests identificeren binnen enkele minuten besmet drinkwater, geen weken
- Antarctische ijsplaten kwetsbaar voor plotselinge door smeltwater veroorzaakte breuken, zegt studie
- Bliksemstormen minder waarschijnlijk in een opwarmende planeet, studie suggereert:
- VS zegt dat lekkende nucleaire afvalkoepel veilig is; Leiders van de Marshalleilanden geloven het niet
- Biologisch stof meten in de wind
- Studie zegt dat dammen in de Mekong rivier levens kunnen verstoren omgeving
Hoofdlijnen
- Samengestelde gezichten van DNA helpen bij het oplossen van koude gevallen
- Ecologische successie: definitie, types, stadia en voorbeelden
- Een nieuw type robotmicroscoop
- Wetenschappers ontdekken dat schorpioenen zich richten op hun gif
- Wat gebeurt er met je cellen als je uitgedroogd bent?
- Een nieuwe strategie die door Helicobacter pylori wordt gebruikt om mitochondriën aan te pakken
- Dakgoten wemelen van onopvallend leven
- Van renpaarden tot bananen:het belang van bioveiligheid
- Cambodja neemt lading ivoor in beslag, verborgen in holle boomstammen
- Op pijnboomsap gebaseerd plastic:een potentiële gamechanger voor de toekomst van duurzame materialen

- Onderzoekers ontrafelen meer mysteries van metallische waterstof

- Chemici ontwikkelen een methode om verbindingen voor geneesmiddelen te synthetiseren
- Video:Hoe wordt leer gemaakt?

- Onderzoekers kweken het meest levensechte bot tot nu toe uit geweven cellen


 Wat zijn de fysieke kenmerken van de Atlantische kustvlakten?
Wat zijn de fysieke kenmerken van de Atlantische kustvlakten? - Nieuw tijdperk in luchtkwaliteitsmonitoring een stap verwijderd
- Lab maakt geleidende 3D-koolstofblokken die kunnen worden gevormd voor toepassingen
- Gezond verstand in robots is niet zo gebruikelijk, maar dit Pictionary-achtige spel kan daar verandering in brengen
- Hoog gemiddeld uitgangsvermogen bereikt in PAPS-lasersysteem met fotokathodeaandrijving
- Een nieuwe methode om de voorspellingen van de intensiteit van tropische cyclonen te verbeteren
- Activisten willen brug over snelweg bouwen om wilde dieren veilig over te steken
- Hoe maak je een piramide voor een schoolproject
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
