Wetenschap
Wetenschappers produceren eerste open source all-atom-modellen van COVID-19 spike-eiwit
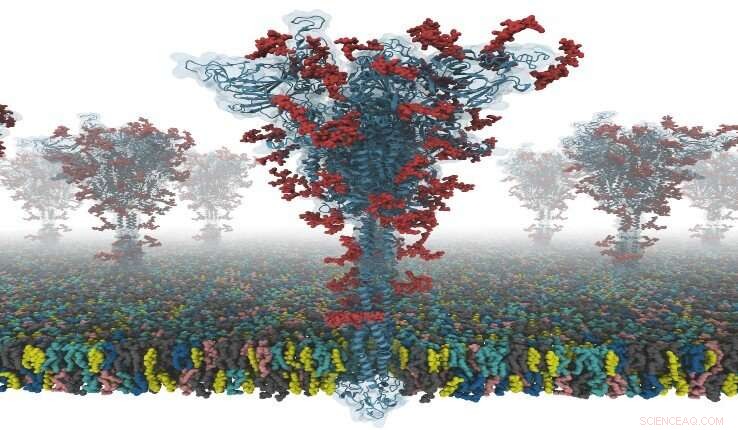
Een model van een S-eiwit. Krediet:Dr. Yeolkyo Choi/Lehigh
Het virus SARS coronavirus 2 (SARS-CoV-2) is de bekende veroorzaker van coronavirusziekte 2019 (COVID-19). Het "spike"- of S-eiwit vergemakkelijkt het binnendringen van virussen in gastheercellen.
Nu heeft een groep onderzoekers van de Seoul National University in Zuid-Korea, Universiteit van Cambridge in het VK, en Lehigh University in de VS, hebben samengewerkt om de eerste open-source all-atom-modellen van een S-eiwit van volledige lengte te produceren. De onderzoekers zeggen dat dit van bijzonder belang is omdat het S-eiwit een centrale rol speelt bij het binnendringen van virussen in cellen, waardoor het een belangrijk doelwit is voor de ontwikkeling van vaccins en antivirale geneesmiddelen.
De details zijn te vinden in een paper, "Ontwikkeling van een volledig geglycosyleerd SARS-CoV-2 Spike-eiwitmodel van volledige lengte in een viraal membraan" zojuist online gepubliceerd in The Journal of Physical Chemistry B .
Deze videodemo illustreert hoe je dit membraansysteem kunt bouwen op basis van hun SARS-CoV-2 S-eiwitmodellen. Het modelbouwprogramma is open access en is te vinden op de startpagina van CHARMM-GUI door op de COVID-19-archieflink te klikken, of door op de archieflink in de kop te klikken, dan de COVID-19 Proteins-link in de linkerzijbalk.
Ontwikkeld door Wonpil Im, een professor in de afdeling Biologische Wetenschappen en Bioengineering van Lehigh University, CHARMM-GUI (GUI =graphical user interface) is een programma dat complexe biomoleculaire systemen eenvoudig simuleert, precies en snel. Im beschrijft het als een 'computationele microscoop' waarmee wetenschappers interacties op moleculair niveau kunnen begrijpen die op geen enkele andere manier kunnen worden waargenomen. Meer informatie over CHARMM-GUI vind je in deze video.
"Onze modellen zijn de eerste volledig geglycosyleerde SARS-CoV-2 spike (S)-eiwitmodellen van volledige lengte die beschikbaar zijn voor andere wetenschappers, " zegt Im. "Ik had het geluk om samen te werken met Dr. Chaok Seok van Seoul National University in Korea en Dr. Tristan Croll van University of Cambridge in het VK. Ons team bracht dagen en nachten door om deze modellen zeer zorgvuldig te bouwen van de bekende cryo- EM-structuurdelen. Modelleren was een hele uitdaging omdat er veel regio's waren waar eenvoudige modellering geen modellen van hoge kwaliteit opleverde."
Wetenschappers kunnen de modellen gebruiken om innovatief en nieuw simulatieonderzoek uit te voeren voor de preventie en behandeling van COVID-19, volgens Im.
De S-eiwitstructuur werd bepaald met cryo-EM met de RBD omhoog (PDB ID:6VSB), en met de RBD omlaag (VOB ID:6VXX). Maar, dit model heeft veel ontbrekende resten. Dus, ze hebben eerst de ontbrekende aminozuurresiduen gemodelleerd, en dan andere ontbrekende domeinen. In aanvulling, ze modelleerden alle potentiële glycanen (of koolhydraten) die aan het S-eiwit zijn gehecht. Deze glycanen voorkomen de herkenning van antilichamen, waardoor het moeilijk is om een vaccin te ontwikkelen. Ze bouwden ook een viraal membraansysteem van een S-eiwit voor simulatie van moleculaire dynamica.
 Oplosmiddelen van de volgende generatie vangen koolstof op met de helft van de energie
Oplosmiddelen van de volgende generatie vangen koolstof op met de helft van de energie- Aluminium en water gebruiken om schone waterstofbrandstof te maken
- Het mengen van zijde met polymeren kan leiden tot betere biomedische implantaten
- Welke elementen kunnen Cobalt combineren?
- Deconstructie van het superfood dat de honingbijhiërarchie bepaalt
- Onderzoek suggereert dat het eten van bonen in plaats van rundvlees de broeikasgassen sterk zou verminderen
- Onderzoekers wenden zich tot oesters als schildwachten voor het opsporen van vervuiling
- Luchtkwaliteit is de grootste bedreiging voor het milieu voor de volksgezondheid, EPI-rapport toont
- Wat veroorzaakte de ijstijden? Kleine oceaanfossielen bieden belangrijk bewijs
- Vulkanische groei cruciaal voor de vorming van Panama
Hoofdlijnen
- Lemur-studie benadrukt de rol van voeding bij het vormgeven van het darmmicrobioom
- Conserveringsonderzoek gebruikt kleine loopbanden om het uithoudingsvermogen van de zeeschildpadden te testen
- Linnaean-classificatie: definitie, niveaus en voorbeelden (met grafiek)
- Het korstmos dat zijn voortplantingsstrategie verandert afhankelijk van het klimaat
- Onderzoek opent poorten voor betere gerichte medicijnen
- Gerst is de smaak van de maand, aangezien nieuwe studie het eeuwenoude brouwdebat beslecht
- Gebruik van microscopen in de wetenschap
- Burgerwetenschappers ontdekken zes nieuwe soorten kevers in Borneo
- Wat is een zuivere eigenschap en een hybride eigenschap?
- Onderzoekers ontwerpen nieuwe biomimetische diamantfilm tegen biofouling

- Hoe interstitiële ordening invloed heeft op hogesterktestaal

- Een nieuw proces om metallurgische slakken te recyclen

- Verankerd door een dichte buurt:wat voorkomt dat cellen afdwalen?

- Elektrisch neutraal radicaal:een sterk chemisch reductiemiddel bij blootstelling aan licht


 Virgin Galactic gaat naar de beurs en leidt race voor ruimtetoerisme
Virgin Galactic gaat naar de beurs en leidt race voor ruimtetoerisme- Wetenschappers stellen 3D-grafeenachtige hyper-honingraatstructuren voor
- Google Assistent binnenkort op bijna 1 miljard apparaten, bedrijf zegt op CES 2019
- De Melkweg zou het leven van ster tot ster kunnen verspreiden
- Satellieten tonen aan dat orkaan Katia niet veel beweegt
- Canada moet zijn CO2-belasting verdubbelen om de emissiedoelstelling te halen:rapport
- Flexibele organische fotovoltaïsche cellen met in-situ niet-thermische fotoreductie van spin-gecoate grafeenoxide-elektroden
- Nieuw ontwerp voor nanodeeltjes die energiezuinig licht absorberen, hoogenergetisch licht uitzenden, kan worden gebruikt in biologische beeldvorming
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway | Italian |

-
Wetenschap © https://nl.scienceaq.com
