Wetenschap
Volgorde van structuur voorspellen
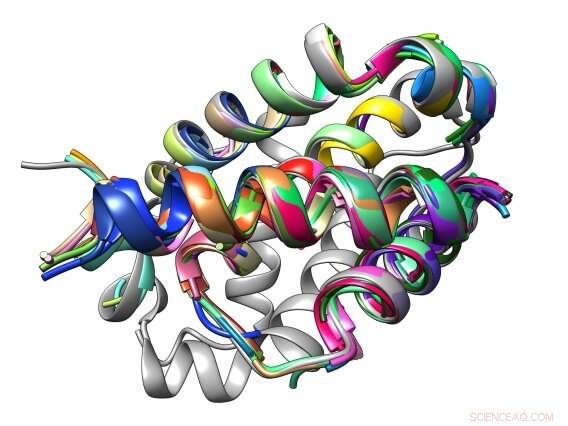
De bindingsinterface tussen een peptide en zijn Bcl-2-eiwitdoelwit is samengesteld uit gemeenschappelijke structurele motieven die bekend staan als TERM's. Krediet:Sebastian Swanson en Avi Singer
Een manier om ingewikkelde biologische systemen te onderzoeken, is door te voorkomen dat hun componenten op elkaar inwerken en te kijken wat er gebeurt. Met deze methode kunnen onderzoekers cellulaire processen en functies beter begrijpen, het vergroten van alledaagse laboratoriumexperimenten, diagnostische testen, en therapeutische interventies. Als resultaat, er is veel vraag naar reagentia die interacties tussen eiwitten belemmeren. Maar voordat wetenschappers snel hun eigen aangepaste moleculen kunnen genereren die daartoe in staat zijn, ze moeten eerst de gecompliceerde relatie tussen sequentie en structuur ontleden.
Kleine moleculen kunnen gemakkelijk cellen binnendringen, maar het grensvlak waar twee eiwitten aan elkaar binden is vaak te groot of mist de kleine holtes die nodig zijn voor deze moleculen om zich te richten. Antilichamen en nanolichamen binden aan langere stukken eiwit, waardoor ze beter geschikt zijn om eiwit-eiwit interacties te belemmeren, maar hun grote omvang en complexe structuur maken ze moeilijk af te leveren en onstabiel in het cytoplasma. Daarentegen, korte stukjes aminozuren, bekend als peptiden, zijn groot genoeg om lange stukken eiwit te binden, terwijl ze toch klein genoeg zijn om cellen binnen te dringen.
Het Keating-lab van het MIT Department of Biology werkt hard aan het ontwikkelen van manieren om snel peptiden te ontwerpen die eiwit-eiwitinteracties met Bcl-2-eiwitten kunnen verstoren, die de groei van kanker bevorderen. Hun meest recente aanpak maakt gebruik van een computerprogramma genaamd dTERMen, ontwikkeld door Keating lab-alumnus, Gevorg Grigoryan Ph.D. '07, momenteel universitair hoofddocent computerwetenschappen en adjunct universitair hoofddocent biologische wetenschappen en scheikunde aan het Dartmouth College. Onderzoekers voeden het programma eenvoudig hun gewenste structuren, en het spuugt aminozuursequenties uit voor peptiden die in staat zijn om specifieke eiwit-eiwitinteracties te verstoren.
"Het is zo'n eenvoudige manier om te gebruiken, " zegt Keating, een MIT-professor biologie en senior auteur van de studie. "In theorie, je zou elke structuur kunnen invoeren en een reeks kunnen oplossen. In onze studie, het programma kwam met nieuwe reekscombinaties die niet lijken op alles wat in de natuur te vinden is - het leidde een volledig unieke manier af om het probleem op te lossen. Het is opwindend om nieuwe gebieden van het sequentie-universum te ontdekken."
Voormalig postdoc Vincent Frappier en Justin Jenson Ph.D. '18 zijn co-eerste auteurs van de studie, die verschijnt in het laatste nummer van Structuur .
Hetzelfde probleem, andere aanpak
jenson, voor zijn deel, heeft de uitdaging aangepakt om peptiden te ontwerpen die binden aan Bcl-2-eiwitten met behulp van drie verschillende benaderingen. De op dTERMen gebaseerde methode, hij zegt, is verreweg de meest efficiënte en algemene die hij tot nu toe heeft geprobeerd.
Standaardbenaderingen voor het ontdekken van peptideremmers omvatten vaak het modelleren van hele moleculen tot aan de fysica en chemie achter individuele atomen en hun krachten. Andere methoden vereisen tijdrovende screening voor de beste bindende kandidaten. In beide gevallen, het proces is zwaar en het slagingspercentage is laag.
dTERMen, daarentegen, vereist geen fysica of experimentele screening, en maakt gebruik van gemeenschappelijke eenheden van bekende eiwitstructuren, zoals alfa-helices en bètastrengen - tertiaire structurele motieven of "TERM's" genoemd - die zijn verzameld in verzamelingen zoals de Protein Data Bank. dTERMen haalt deze structurele elementen uit de databank en gebruikt ze om te berekenen welke aminozuursequenties een structuur kunnen aannemen die in staat is om specifieke eiwit-eiwit interacties te binden en te onderbreken. Het duurt één dag om het model te bouwen, en slechts enkele seconden om duizend sequenties te evalueren of een nieuw peptide te ontwerpen.
"dTERMen stelt ons in staat sequenties te vinden die waarschijnlijk de bindingseigenschappen hebben waarnaar we op zoek zijn, in een robuuste, efficiënt, en algemene wijze met een hoog succespercentage, " zegt Jenson. "De eerdere benaderingen hebben jaren geduurd. Maar met dTERMen, we gingen in een paar weken van constructies naar gevalideerde ontwerpen."
Van de 17 peptiden die ze bouwden met behulp van de ontworpen sequenties, 15 gebonden met native-achtige affiniteit, het verstoren van Bcl-2-eiwit-eiwit-interacties die notoir moeilijk te richten zijn. In sommige gevallen, hun ontwerpen waren verrassend selectief en gebonden aan een enkel Bcl-2-familielid boven de anderen. De ontworpen sequenties weken af van bekende sequenties die in de natuur worden gevonden, wat het aantal mogelijke peptiden aanzienlijk verhoogt.
"Deze methode laat een zekere mate van flexibiliteit toe, " zegt Frappier. "dTERMen is beter bestand tegen structurele verandering, waardoor we nieuwe soorten structuren kunnen verkennen en onze portefeuille van potentiële bindende kandidaten kunnen diversifiëren."
Het sequentie-universum onderzoeken
Gezien de therapeutische voordelen van het remmen van de Bcl-2-functie en het vertragen van tumorgroei, het Keating-lab is al begonnen met het uitbreiden van hun ontwerpberekeningen naar andere leden van de Bcl-2-familie. Ze zijn van plan om uiteindelijk nieuwe eiwitten te ontwikkelen die structuren aannemen die nog nooit eerder zijn gezien.
"We hebben nu genoeg voorbeelden gezien van verschillende lokale eiwitstructuren dat computationele modellen van sequentie-structuurrelaties direct kunnen worden afgeleid uit structurele gegevens, in plaats van telkens opnieuw te moeten worden ontdekt vanuit atomistische interactieprincipes, " zegt Grigoryan, de maker van dTERMen. "Het is enorm opwindend dat dergelijke op structuur gebaseerde inferentie werkt en nauwkeurig genoeg is om robuust eiwitontwerp mogelijk te maken. Het biedt een fundamenteel ander hulpmiddel om de belangrijkste problemen van structurele biologie aan te pakken - van eiwitontwerp tot structuurvoorspelling."
Frappier hoopt op een dag het hele menselijke proteoom computationeel te kunnen screenen, met behulp van methoden zoals dTERMen om kandidaat-bindende peptiden te genereren. Jenson suggereert dat het gebruik van dTERMen in combinatie met meer traditionele benaderingen voor het herontwerpen van sequenties een toch al krachtig hulpmiddel zou kunnen versterken, onderzoekers in staat stellen deze gerichte peptiden te produceren. Ideaal, hij zegt, op een dag kan het ontwikkelen van peptiden die je favoriete eiwit binden en remmen net zo eenvoudig zijn als het uitvoeren van een computerprogramma, of zo routinematig als het ontwerpen van een DNA-primer.
Volgens Keating, hoewel die tijd nog in de toekomst ligt, "onze studie is de eerste stap naar het aantonen van deze capaciteit op een probleem van bescheiden omvang."
Dit verhaal is opnieuw gepubliceerd met dank aan MIT News (web.mit.edu/newsoffice/), een populaire site met nieuws over MIT-onderzoek, innovatie en onderwijs.
 Visserijgeschiedenis wijst op substantiële achteruitgang voor belangrijke soorten
Visserijgeschiedenis wijst op substantiële achteruitgang voor belangrijke soorten- EU heroverweegt hulp voor civiele bescherming na branden in Portugal
- NASA's infraroodanalyse van tropische storm Sebastien ziet windschering
- Experts waarschuwen voor dode zone in Chesapeake Bay tegen vervuiling
- Vlak, trein, of auto? De klimaatimpact van transport is ingewikkeld
Hoofdlijnen
- De reden voor incubatie bij verschillende temperaturen in de microbiologie
- Twee nieuwe soorten schaaldieren ontdekt op Galicische zeebodem
- Nieuwe sorghumcultivars kunnen duizenden liters ethanol produceren
- Wat is één reden waarom de classificatie van protisten in één koninkrijk moeilijk is?
- Op weg naar pesticidebewaking
- High School Biology Topics
- Drie voorbeelden van protisten met wetenschappelijke namen
- Hoe kunnen kinderen van dezelfde ouders er zo verschillend uitzien?
- Woedend debat:houdt het ruimen van wolven stroperij tegen?
- Nieuw katalytisch effect ontdekt voor de productie van galliumoxide
- Bacteriën eten broeikasgas met een kant van eiwit

- Magnetisch gestuurd, op hydrogel gebaseerde slimme transformatoren
- Onderzoekers realiseren 4D-geprint materiaal

- Nieuwe röntgenmethode heeft ingrijpende gevolgen voor de ontwikkeling van levensreddende medicijnen

 Een ruimtevenster naar opwindende wetenschap
Een ruimtevenster naar opwindende wetenschap- Wat zijn de twee hoofdfasen van celdeling?
- Europa vuren worden erger, zelfs als de klimaatdoelen worden gehaald:studie
- Hellingstabiliteitsmodel kan aardverschuivingen helpen voorspellen om gemeenschappen te beschermen, Red levens
- Optisch apparaat ontleedt een straal in een Cartesiaans raster van identieke Gaussische vlekken
- De Noordelijke IJszee was bedekt met een plankijs en gevuld met zoet water
- Dataschandaal bedreigt Zuckerberg-visie voor Facebook
- Wetenschappers vinden ijzersneeuw in de kern van de aarde
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
