Wetenschap
Minder is meer als het gaat om het voorspellen van de geleidbaarheid van moleculen

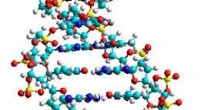
UChicago afgestudeerde student Manas Sajjan, links, en prof. David Mazziotti, hebben een model dat een molecuul weergeeft waarop ze een betere benadering hebben getest om geleidbaarheid te voorspellen. Krediet:Jean Lachat/Universiteit van Chicago
Hoe kleiner en slimmer telefoons en apparaten worden, hoe groter de noodzaak om kleinere circuits te bouwen. Vooruitstrevende wetenschappers in de jaren zeventig suggereerden dat circuits konden worden gebouwd met behulp van moleculen in plaats van draden, en in de afgelopen decennia is die technologie realiteit geworden.
Het probleem is, sommige moleculen hebben bijzonder complexe interacties die het moeilijk maken om te voorspellen welke van hen goed zouden kunnen dienen als miniatuurcircuits. Maar een nieuw artikel van twee chemici van de Universiteit van Chicago presenteert een innovatieve methode die de rekenkosten verlaagt en de nauwkeurigheid verbetert door interacties tussen elektronenparen te berekenen en die te extrapoleren naar de rest van het molecuul.
"De huidige modellen hebben de neiging om geleiding te overschatten, maar onze theorie overtreft de traditionele modellen met wel één tot twee ordes van grootte, " zei prof. David Mazziotti, die co-auteur was van het artikel, gepubliceerd op 17 mei in Nature's Communicatiechemie .
Alles, van betere computerchips en batterijen tot groenere manieren om chemicaliën te produceren, hangt af van het ontdekken van nieuwe soorten chemicaliën en materialen, en wetenschappers kijken steeds vaker naar computers om efficiënter naar nieuwe combinaties te zoeken. In plaats van permutaties één voor één te proberen, ze kunnen modellen uitvoeren die de beste opties voorspellen.
Maar het is een delicate kunst, omdat deze berekeningen in veel gevallen schrikbarend snel rekentijd kunnen kosten. In moleculen met veel interagerende elektronen, "je kunt heel snel eindigen met de rekengrootte die exponentieel toeneemt met de grootte van het molecuul, ' zei Mazziotti.
Mazziotti en afgestudeerde student Manas Sajjan wilden vereenvoudigen, het creëren van een methode voor het voorspellen van moleculaire geleidbaarheid die de interactie tussen twee elektronen gebruikt om alle interacties weer te geven. "Om een voorbeeld te noemen, voor een bepaald molecuul vereist de traditionele methode mogelijk berekeningen met 1024 variabelen, terwijl de onze 109 variabelen heeft - een biljard minder variabelen, " zei Sajjan. Dat is het verschil tussen een probleem waarvoor je een supercomputer nodig hebt en een probleem dat je op een laptop kunt oplossen.
Deze keuze maakt een ongebruikelijke maar krachtige aanpak mogelijk. Bestaande theorieën voor moleculaire geleidbaarheid kennen een vast aantal spanningen toe die op het molecuul worden toegepast om een getal te voorspellen voor de stroom die er dan doorheen zou kunnen stromen. Sajjan en Mazziotti hebben dit paradigma op zijn kop gezet. Ze hebben eerst de stroom gerepareerd, en vervolgens de spanning berekend. Dit blijkt veel nauwkeuriger te zijn:toen ze hun methode controleerden met een bekend molecuul, ze zagen dat het een-op-twee beter presteerde dan traditionele methoden.
"Wat belangrijk is, is dat het echt rigoureus is. Zelfs met de geleiding is er nog steeds een één-op-één mapping met het veel-elektronensysteem, " zei Mazziotti. Het proces om ervoor te zorgen dat het twee-elektronensysteem nog steeds het veel-elektronensysteem vertegenwoordigt, is een zeer uitdagend probleem dat al 50 jaar bestaat, maar hij zei dat het de strijd waard was.
"Bijna alle grote problemen die mensen proberen op te lossen, hebben betrekking op het werken met materialen die moeilijk te onderzoeken zijn met traditionele methoden, " zei hij. "Als we de geleidbaarheid beter kunnen voorspellen, we kunnen effectiever betere moleculen en materialen ontwerpen."
 Steenkool onthult een verfijnde kant:vuile koolstof kan worden gebruikt om een verscheidenheid aan nuttige apparaten te maken
Steenkool onthult een verfijnde kant:vuile koolstof kan worden gebruikt om een verscheidenheid aan nuttige apparaten te maken- De nieuwste:Nobelprijswinnaar zoals Google Earth voor moleculen
- Neutronen onthullen snelle translatie van methaan op het grensvlak van twee clathraatstructuren
- Apparaat voor het gelijktijdig meten van gassen die de kwaliteit van de binnenlucht verminderen
- CO-biosynthese vereist voor de assemblage van de actieve plaats in NiFe-hydrogenase
Hoofdlijnen
- Wanneer citroenen je leven geven:Herpetofauna-aanpassing aan citrusboomgaarden in Belize
- Hoe is DNA geordend om in een cel te passen?
- Wat is het verschil tussen ras en etniciteit?
- Lichaamsdelen en functies
- Zijn psychische aandoeningen genetisch bepaald?
- Mannetjes passen de snelheid van het sperma snel aan om rivalen te verslaan, studie vondsten
- Een 3D-model van een dier of plant maken Cell
- Celstructuurdefinities
- Onderzoek werpt nieuw licht op hoe organismen energie gebruiken in een menigte




 Verlaten akkerland moet biobrandstoffen produceren
Verlaten akkerland moet biobrandstoffen produceren- Californië bestrijdt grootste brand ooit:acht doden in het westen van de VS
- Een nieuwe formule voor het creëren van chemische reacties - met koolhydraten
- Doorbraak in elektrolyse kan het waterstofvraagstuk oplossen
- Veranderingen in suikerachtige moleculen aan het oppervlak helpen kanker uitzaaien
- Recordbrekend zwak satellietstelsel van de Melkweg ontdekt
- Hoe nanotechnologie regeneratieve geneeskunde kan bevorderen
- Duitsland geeft 3 terug, 000 jaar oude houten Olmeken bustes naar Mexico
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
