Wetenschap
Hoe computers op zoek zijn naar drugs van de toekomst

Krediet:Universiteit van Californië, San Francisco
De ontdekking van geneesmiddelen doet denken aan beelden van witte laboratoriumjassen en pipetten, maar toen Henry Lin, doctoraat, onlangs op zoek naar een betere opioïde met minder bijwerkingen, zijn eerste stap was het opstarten van de computers.
Met behulp van een programma genaamd DOCK, hij uploadde een kristalstructuur van de opioïde receptor in de hersenen en kreeg toegang tot een virtuele bibliotheek van 3 miljoen verbindingen die zich zouden kunnen binden aan een chemisch "zakje" op de receptor. De meeste medicijnen – van antibiotica tot antidepressiva – werken door zich te binden aan specifieke plaatsen op eiwitten, maar om effectief te zijn, ze moeten precies goed passen.
Het programma draaide elke verbinding rond, rekening gehouden met de flexibiliteit van de verschillende aanhangsels, en na het testen van gemiddeld 1,3 miljoen configuraties per verbinding - gerangschikt op hun bindingspotentieel. Het proces, draaien op computers die zijn aangesloten op krachtige processors, duurde ongeveer twee weken.
Een afstudeerder destijds, Lin werkte samen met zijn adviseur Brian Shoichet, doctoraat, hoogleraar farmaceutische chemie aan de UC San Francisco School of Pharmacy, en Aashish Manglik, doctoraat, van Stanford University om door de top 2 te kammen, 500 verbindingen voor aanvullende factoren en 23 geselecteerd voor experimenteel testen in levende cellen - cue laboratoriumjassen en pipetten.
Meer en meer, onderzoekers wenden zich tot virtuele experimenten voor de eerste stappen van de ontwikkeling van geneesmiddelen. Met steeds snellere computers, de vroege en grotendeels trial-and-error-fase van de ontwikkeling van geneesmiddelen kan worden teruggebracht tot een kwestie van dagen, en met steeds groter wordende online bibliotheken van verbindingen, drugsschermen kunnen omvatten, letterlijk, alle bekende chemie ter wereld.
Sterke punten en beperkingen
Onderzoekers zijn voorzichtig met het potentieel van computationele geneesmiddelenontdekking - slechts een klein deel van de veelbelovende verbindingen werkt echt wanneer ze in het echte leven worden getest - maar ze zeggen dat een van de kracht ervan is om volledig nieuwe verbindingen als kandidaat-geneesmiddelen te onthullen.
Shoichet is gespecialiseerd in een populaire rekenmethode die bekend staat als moleculaire docking. "Waar docking past, is in vroeg ontdekkingsonderzoek, bij het vinden van nieuwe vertrekpunten, " hij zei.
De zoektocht van zijn team naar het nieuwe opioïde illustreert zowel de sterke punten als de beperkingen van computationele medicijnontdekking.
In feite, de initiële opioïde-kandidaten die werden geïdentificeerd door middel van moleculaire koppeling presteerden slechts bescheiden in experimentele tests. "Nog altijd, de activiteit die ze hadden was zeer reproduceerbaar en de moleculen waren zeer nieuw, vooruitstrevende nieuwe biologie, ' zei Shoichet.
Het team legde nog een ronde verbindingen met vergelijkbare structuren aan en testte de topscorers. Met medewerkers van de Universiteit van North Carolina, Chapel Hill en Friedrich Alexander University in Duitsland, ze identificeerden de meest krachtige verbinding en optimaliseerden de farmacologie ervan met computergestuurde synthetische uitwerking.
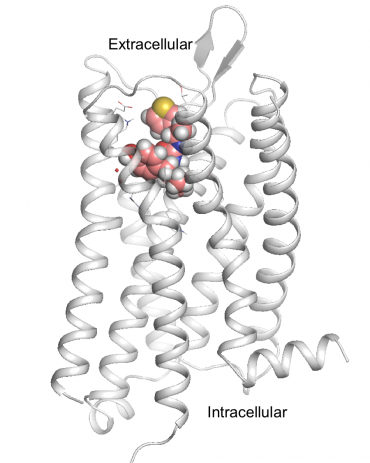
PZM21, de nieuwe, veiliger opioïde kandidaat-medicijn, wordt weergegeven als gekoppeld aan de morfinereceptor van de hersenen, de mu-opioïde receptor. Krediet:Anat Levit
Dat winnende mengsel, genaamd PZM21, is chemisch anders dan in het huidige gebruik en is mogelijk niet gevonden via meer traditionele methoden. Het is een volledig computationeel ontworpen verbinding die krachtiger is dan morfine. In muizen, het blokkeerde efficiënt pijn zonder de gebruikelijke bijwerkingen van ademhalingssuppressie en constipatie en leek zelfs minder verslavend te zijn.
Docking is geen wondermiddel, maar het is een krachtig startpunt geworden voor de lange, interdisciplinair proces van medicijnontwikkeling. Een van de belangrijkste bijdragen was de proteaseremmers die ertoe hebben bijgedragen dat hiv een behandelbare ziekte is geworden. Onderzoekers gebruiken docking ook om kandidaat-geneesmiddelen te screenen voor de behandeling van borstkanker, hepatitis C, hypertensie, stafylokokken, het SARS-virus en griep.
Technologie pionierde bij UCSF
Moleculair docking werd drie decennia geleden ontwikkeld door een jonge UCSF fysisch chemicus genaamd Tack Kuntz, doctoraat, nu emeritus hoogleraar aan de School of Pharmacy. Toen Kuntz begin jaren zeventig op de campus arriveerde, de traditionele benadering van het ontdekken van geneesmiddelen had nog steeds de overhand.
Zoals Kuntz het beschreef, het proces was gebaseerd op toeval en heel weinig theorie:"Je gaat op zoek naar nieuwe natuurlijke verbindingen en brengt ze terug om te testen in een laboratorium. Voeg gewoon chemicaliën samen met een organisme en kijk wat er gebeurt."
Farmaceutische chemici hebben nauwelijks nagedacht over de moleculaire details van de interactie van geneesmiddelen met het lichaam. Veel medicijnen, inclusief de eerste antibiotica, bij toeval ontdekt, maar Kuntz, gezien het nieuwe moleculaire begrip het veld van de biologie overspoelde, vond dat het tijd was voor een soortgelijke update in de farmacologie.
"De op doelen gebaseerde kijk op biologie - dat je biologie kunt begrijpen door onafhankelijke eiwitten en genproducten - had het al overgenomen, maar de farmacologie liep een decennium achter, " zei Shoichet, die in de jaren tachtig een afgestudeerde student was in het laboratorium van Kuntz.
Kuntz en zijn collega's begonnen te werken aan een meer rationele benadering van het ontwerpen van geneesmiddelen, waarbij ze probeerden verbindingen te identificeren die op specifieke receptoren op eiwitten konden passen, zoals het vinden van het ontbrekende stukje van een legpuzzel. 1982, ze publiceerden een paper waarin het eerste moleculaire koppelingsprogramma werd beschreven dat "geometrisch haalbare uitlijningen van liganden en receptoren met een bekende structuur zou kunnen onderzoeken".
Kuntz stuurde 10, 000 exemplaren van dat eerste koppelingsprogramma aan onderzoekers in het hele land. Spoedig, andere onderzoekers ontwikkelden soortgelijke computerprogramma's en de opwinding verspreidde zich snel buiten de academische wereld. Tegen de jaren negentig, elk groot farmaceutisch bedrijf had een computationele eenheid voor het ontdekken van geneesmiddelen geopend.
Een idee inhalen
Ondanks het aanvankelijke enthousiasme, echter, computationele medicijnontdekking leidde niet tot snelle resultaten. Het idee van Kuntz was zijn tijd ver vooruit. Het zou tientallen jaren van stapsgewijze vooruitgang in de moleculaire biologie vergen, beeld- en computertechnologie, voordat computationele medicijnontdekking zijn belofte kon waarmaken.

Tack Kuntz, doctoraat, en zijn collega's publiceerden in 1982 een paper waarin het eerste moleculaire koppelingsprogramma werd beschreven dat "geometrisch haalbare uitlijningen van liganden en receptoren met een bekende structuur zou kunnen onderzoeken". Krediet:Universiteit van Californië, San Francisco
Een belangrijke beperking in de jaren negentig was het ontbreken van bekende eiwitstructuren. Zonder deze, er waren maar weinig doelwitten om drugs te vinden. In de decennia daarna, duizenden eiwitstructuren van mogelijke medicijndoelen zijn onthuld door röntgenkristallografie en nucleaire magnetische resonantiebeeldvorming.
De ontdekking van de nieuwe opioïde kandidaat, bijvoorbeeld, was alleen mogelijk vanwege de recent vastgestelde kristalstructuren van G-eiwit-gekoppelde receptoren, een familie van eiwitten die de opioïde receptor omvat.
Virtuele bibliotheken van verbindingen zijn ook exponentieel gegroeid. In 1991, een database kan 55 bevatten, 000 verbindingen; nu bevatten ze tientallen miljoenen. "De reikwijdte van de chemie die we bemonsteren, is ongeveer in hetzelfde tempo gestegen als de wet van Moore, "Zei Shoichet. "Er is een onverzadigbare honger naar meer en meer moleculen."
De huidige dockingprogramma's zijn in staat om de interacties op atomair niveau tussen een medicijn en zijn doelwit realistisch te modelleren, maar enkele lastige details - zoals hoe atomaire krachten veranderen wanneer een medicijnmolecuul water op de bindingsplaats verdringt - blijven voortdurende uitdagingen in het veld.
Beloften en bewijzen
Moleculaire docking is niet de enige vorm van computergebaseerd medicijnontwerp. Aan het UCSF Institute for Computational Health Sciences (ICHS), tientallen onderzoekers onderzoeken talloze computationele methoden om medisch onderzoek vooruit te helpen.
Michaël Keizer, doctoraat, een lid van ICHS en een assistent-professor aan het Institute of Neurodegeneratieve Ziekten, bestudeert medicijnen die veel moleculaire doelen tegelijk raken, alsof je een akkoord aanslaat in plaats van een enkele noot. Deze multi-target actie werd lang gezien als de oorzaak van onbedoelde bijwerkingen, maar kan ook worden gericht op de behandeling van complexe ziekten.
Pas in het begin van de jaren 2000 kwamen onderzoekers tot de erkenning dat veel bestaande medicijnen via meer dan één doelwit werken:antipsychotica, bijvoorbeeld, die zowel serotonine- als dopaminereceptoren raken. Ze zijn nu opzettelijk medicijnen aan het ontwerpen om dit te doen.
"Voor sommige ziekten die nog geen behandelingen hebben, misschien is het omdat er geen enkel eiwit is dat je aan of uit hoeft te zetten; wat als het medicijn in plaats daarvan meerdere doelen moet raken?" zei Keizer, die een afgestudeerde student van Shoichet was.
In zijn laboratorium, Keizer gebruikt computationele methoden om chemische patronen te identificeren tussen geneesmiddelen die aan dezelfde set doelen binden en nieuwe verbindingen te vinden die overeenkomen met de farmacologie. Deze computationele benadering kan overeenkomsten tussen verbindingen herkennen die meer conventionele analyses zouden missen. Keizer kijkt nu naar kunstmatige intelligentietechnologie, bekend als diep leren, voor een nog betere patroonherkenning.
Zelfs als computationele methoden populair worden, hun bewijs is nog steeds in de echte wereld - in cellen, diermodellen, en uiteindelijk in de kliniek. "Een tijdje was het gebruikelijk om artikelen te publiceren met voorspellingen over de activiteiten van een klein molecuul, maar geen daadwerkelijke testen van deze voorspellingen, omdat de experimenten om dit te doen duur waren, moeilijk of esoterisch, ' zei Keizer.
Nu de noodzaak tot samenwerking duidelijk is geworden, de samenwerking tussen computationele voorspelling en natte lab-experimenten is het afgelopen decennium merkbaar versterkt, zei Keizer. "Ten slotte, hoe kun je je voorspellingen verbeteren als je niet zeker weet welke fout zijn?"
 De magische boom van Latijns-Amerika komt langzaam weer tot leven
De magische boom van Latijns-Amerika komt langzaam weer tot leven- Klimaatverandering kan leiden tot een algehele toename van de rivierafvoer, maar meer droogtes en overstromingen, studie toont
- Het morele element van klimaatverandering
- Onderzoekers meten koolstofvoetafdruk van waterkrachtcentrales in Canada
- Waarom groeien er maden op vlees?
Hoofdlijnen
- Snelle reactie van Fish op klimaatverandering
- Waarom heeft de evolutie ons niet het vermogen gegeven om zoet water te ruiken?
- Voorbeelden van genetische kenmerken
- Accuplacer-regels
- Atrazine verandert de sex-ratio in Blanchards krekelkikkers
- Zijn mensen echt afstammelingen van apen?
- Wat zijn drie primaire doelen van mitose?
- Openbare bronnen stimuleren de ontdekking van medicijnen en bieden inzicht in de eiwitfunctie
- Bronnen van fouten in gelelektroforese
- Warme melk maakt je slaperig - peptiden kunnen verklaren waarom

- Wetenschappers ontdekken nieuwe benadering om kathodematerialen te stabiliseren

- Lipidenblaasjes vervangen bloed in nieuwe bacterietest
- De rol van hydrofobe moleculen in katalytische reacties

- Wetenschappers ontwikkelen manier om salmonella-infectie in realtime te volgen

 Nieuwe elektronische koeltechnologie om miniaturisatie van kwantumcomputers mogelijk te maken
Nieuwe elektronische koeltechnologie om miniaturisatie van kwantumcomputers mogelijk te maken- Het ontwerp van moleculen automatiseren om de ontwikkeling van geneesmiddelen te versnellen
- De sociale verantwoordelijkheid van Facebook moet privacybescherming omvatten
- Voorgestelde kwantum nano-MRI zou afbeeldingen kunnen genereren met een resolutie op angstrom-niveau
- Nieuwe draagbare sensor volgt vitamine C-niveaus in zweet
- Wanneer T-cellen van ons immuunsysteem actief worden, kleine trekkrachten op moleculair niveau spelen een belangrijke rol
- Het stapelen en draaien van grafeen ontgrendelt een zeldzame vorm van magnetisme
- Wintertarwe haalbaar dekgewas voor Rolling Plains-katoen
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
