Wetenschap
Studie biedt een krachtige computermodelleringsbenadering voor celsimulaties
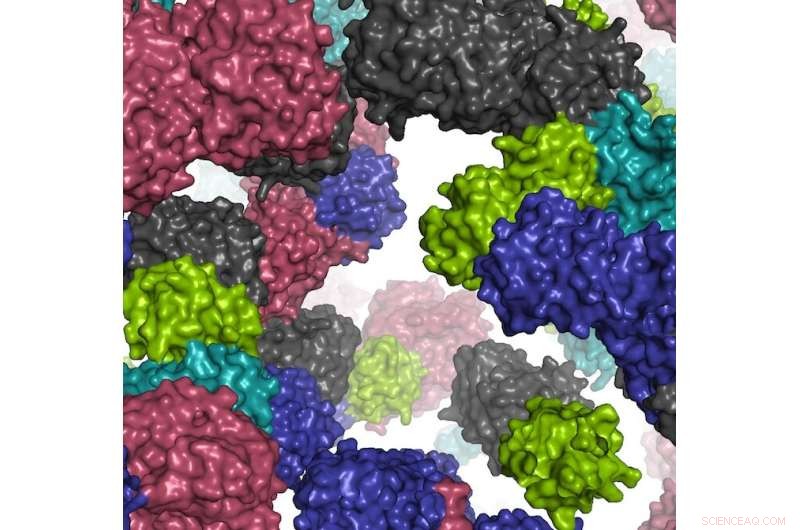
Een fragment van de gesimuleerde celomgeving. Krediet:Ilya Vakser
Een mijlpaalrapport van de Universiteit van Kansas dat deze week verschijnt in de Proceedings of the National Academy of Sciences stelt een nieuwe techniek voor om het moleculaire leven met computers te modelleren.
Volgens hoofdauteur Ilya Vakser, directeur van het Computational Biology Program and Centre for Computational Biology en hoogleraar moleculaire biowetenschappen aan de KU, is het onderzoek naar computermodellering van levensprocessen een belangrijke stap in de richting van het creëren van een werkende simulatie van een levende cel met atomaire resolutie . De vooruitgang belooft nieuwe inzichten in de fundamentele biologie van een cel, evenals een snellere en nauwkeurigere behandeling van menselijke ziekten.
"Het is ongeveer tien- of honderdduizenden keren sneller dan de bestaande atomaire resolutietechnieken", zei Vakser. "Dit biedt ongekende mogelijkheden om fysiologische mechanismen te karakteriseren die nu ver buiten het bereik van computationele modellering liggen, om inzicht te krijgen in cellulaire mechanismen en om deze kennis te gebruiken om ons vermogen om ziekten te behandelen te verbeteren."
Tot nu toe was een grote hindernis bij het modelleren van cellen via de computer de benadering van eiwitten en hun interacties die de kern vormen van cellulaire processen. Tot op heden waren gevestigde technieken voor het modelleren van eiwitinteracties afhankelijk van "eiwitdocking" of "moleculaire simulatie".
Volgens de onderzoekers hebben beide benaderingen voor- en nadelen. Hoewel algoritmen voor het koppelen van eiwitten geweldig zijn voor het bemonsteren van ruimtelijke coördinaten, houden ze geen rekening met de "tijdcoördinaat" of dynamiek van eiwitinteracties. Daarentegen modelleren moleculaire simulaties de dynamiek goed, maar deze simulaties zijn te traag of hebben een lage resolutie.
"Onze proof-of-concept-studie overbrugt de twee modelleringsmethodologieën en ontwikkelt een benadering die ongekende simulatietijdschalen kan bereiken met een resolutie van alle atomen", schreven de auteurs.
Medewerkers van Vakser op het papier waren Sergei Grudinin van de Universiteit van Grenoble Alpes in Frankrijk; Eric Deeds van de Universiteit van Californië-Los Angeles; KU-promovendus Nathan Jenkins en Petras Kundrotas, assistent-onderzoeksprofessor bij het Computational Biology Program van de KU.
Nadat het team had bedacht hoe de voordelen van de twee benaderingen voor eiwitmodellering het beste konden worden gecombineerd, ontwikkelde en codeerde het team een algoritme om de nieuwe simulatie aan te sturen.
"De moeilijkste uitdaging was om het algoritme te ontwikkelen dat het eenvoudige basisidee van de aanpak adequaat weergeeft," zei Vakser.
Maar zodra ze die doorbraak hadden gemaakt, konden ze de nieuwe procedure valideren.
"Het paradigma was eenvoudig - een duidelijke lijn", zei Vakser.
"De bestaande simulatiebenaderingen brengen het grootste deel van de rekentijd door met reizen in gebieden met een lage waarschijnlijkheid of hoge energie van het systeem. We weten allemaal waar deze gebieden zijn. In plaats daarvan was het idee om alleen in de hoge -waarschijnlijkheid, gebieden met lage energie, en om de gebieden met een lage waarschijnlijkheid over te slaan door de overgangssnelheden tussen de staten met een hoge waarschijnlijkheid te schatten. Het paradigma is zo oud als de biomoleculaire modellering zelf en wordt op grote schaal gebruikt sinds het begin van het modelleringstijdperk decennia geleden."
Maar Vakser zei dat tot het nieuwe artikel van zijn team de benadering niet was toegepast op de kinetiek van eiwitinteracties in de cellulaire omgeving, de focus van hun onderzoek.
"Omdat er veel minder staten met een hoge waarschijnlijkheid zijn dan de staten met een lage waarschijnlijkheid, heeft dat ons een enorme winst opgeleverd in de rekensnelheid - tien- tot honderdduizenden keren", zei Vakser. "Dit werd gedaan zonder duidelijk verlies van nauwkeurigheid. Men kan stellen dat de nauwkeurigheid werd bereikt, omdat het simulatieprotocol is gebaseerd op de 'docking'-technieken, die specifiek zijn ontworpen voor het karakteriseren van eiwitassemblages."
De KU-onderzoeker zei dat zijn celsimulatiemethode zou kunnen worden ingezet om de menselijke gezondheid te onderzoeken en ziekten met een nieuw niveau van precisie te behandelen.
"De aanpak kan worden gebruikt om moleculaire routes te bestuderen die ten grondslag liggen aan ziektemechanismen," zei Vakser. "Het kan worden gebruikt om schadelijke effecten van genetische mutaties te bepalen door de veranderde patronen van eiwitassociaties - genetische mutaties veroorzaken veranderingen in de structuur van eiwitten, die op hun beurt de eiwitassociatie beïnvloeden. Of het kan worden gebruikt om doelen te identificeren voor het ontwerpen van geneesmiddelen door het detecteren van kritische elementen in eiwit-associatiepatronen."
Volgens Vakser biedt de nieuwe simulatietechniek veel veelbelovende onderzoeksmogelijkheden om in de toekomst te verkennen.
"Onder hen is het aanpassen van de benadering van eiwitinteracties met nucleïnezuren, RNA en DNA," zei hij. "We willen ook rekening houden met de flexibiliteit van moleculaire vormen, correleren met het zich snel ontwikkelende spectrum van experimentele studies van de cellulaire omgeving en de procedure toepassen op een model van een echte cel - met zijn feitelijke moleculaire componenten bij elkaar gepakt." + Verder verkennen
Wetenschap aan de vooravond van 'transformationeel' begrip van het leven via celmodellering, zeggen onderzoekers
 Verschuiving naar meer intense regens bedreigt historische Italiaanse wijnmakerij
Verschuiving naar meer intense regens bedreigt historische Italiaanse wijnmakerij- NASA-stagiairs helpen het terminatorprobleem op te lossen via GLOBE challenge
- Levenscyclus van een haai
- Oase-effect in stadsparken kan bijdragen aan de uitstoot van broeikasgassen, studie vondsten
- Het vrijkomen van radioactieve deeltjes in Fukushima was significant, zegt nieuw onderzoek
Hoofdlijnen
- Waar komt collageen vandaan?
- DNA-sequentiebepaling: definitie, methoden, voorbeelden
- Haaienbioloog werkt samen met ruimtevaartingenieur om het gedrag van oceanische wittips te ontdekken
- Waarom kan je jezelf niet kietelen?
- Kun je leven zonder zuurstof? Dit dier kan
- Wetenschappers afluisteren onbekende spitssnuitdolfijnen af om te zien hoe diep ze duiken
- Een flitsende gele keelzanger? Daar zijn genen voor
- Onderzoekers ontdekken dat stress tijdens de zwangerschap de grootte van de baby beïnvloedt
- Dysmorfologie
- Vermont ziet hedendaags record voor reproductie van Amerikaanse zeearenden

- Hoe worden genen aan- en uitgezet?

- Verschil tussen Homozygoot en Heterozygoot

- Stadsuitbreiding in Perth die het seksleven van planten beïnvloedt

- Dierenartsen voeren de eerste bekende hersenoperatie uit om hydrocephalus bij pelsrobben te behandelen

 Hoe te testen op zuurgraad
Hoe te testen op zuurgraad- Als werknemers zich machteloos voelen, ze worden paranoïde en agressief
- Een model van de draaivleugel maken
- Disney sluit deal van $ 71 miljard voor entertainmentactiva van Fox
- Hoe ontzilting werkt
- Nieuwe ultrasnelle methode om antibioticaresistentie te bepalen
- Levensduur van plastic zonnecel springt van uren naar 8 maanden
- Emoji onthult witheid als aanjager van technologie
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
