Wetenschap
Ontvouwende adsorptie op metalen nanodeeltjes:stabiliteit verbinden met katalyse
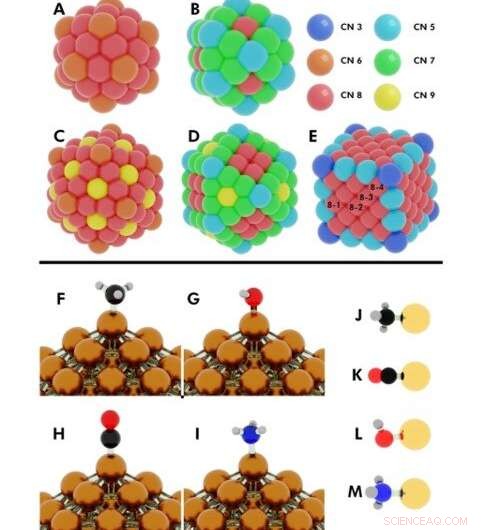
Illustratie van de initiële configuraties voor verschillende uitgevoerde DFT-berekeningen (density functional theory). Boven:Coördinatienummers op (A) 55-atoom icosaëder, (B) 55-atoom kuboctaëder, (C) 147-atoom icosaëder, (D) 147-atoom cuboctaëder, (E) kubus van 172 atomen. Nanodeeltjes (NP's) waarbij meer dan één uniek atoom hetzelfde coördinatienummer (CN) deelt, worden aangegeven met nummers 8-1, 8-2, 8-3, 8-4. Krediet:wetenschappelijke vooruitgang, doi:10.1126/sciadv.aax5101
Metalen nanodeeltjes hebben veel aandacht gekregen vanwege hun toepassingen op diverse gebieden van geneeskunde, katalyse, energie en het milieu. Echter, de fundamentele eigenschappen van adsorptie van nanodeeltjes op een oppervlak moeten nog worden begrepen. James Dean en een interdisciplinair onderzoeksteam van de afdeling Chemical Engineering, in de VS een universeel adsorptiemodel geïntroduceerd om rekening te houden met de structurele kenmerken, metaalsamenstelling en verschillende adsorbaten van nanodeeltjes via machine learning (ML). Het model past op een groot aantal gegevens om adsorptietrends op monometallische en op legeringen gebaseerde nanodeeltjes nauwkeurig te voorspellen. De sjabloon was eenvoudig en leverde snel berekende gegevens voor metalen en adsorbaten. Het onderzoeksteam bracht de adsorptie in verband met stabiliteitsgedrag om het ontwerp van optimale nanodeeltjes voor interessante toepassingen te bevorderen. Het onderzoek is nu gepubliceerd op wetenschappelijke vooruitgang .
Metalen nanodeeltjes (NP's) hebben belangrijke toepassingen in katalyse, variërend van brandstof en chemische productie, tot zonne- en chemische energie. Maar hun stabiliteit en katalytische activiteit vertonen over het algemeen tegengestelde trends, waar zeer actieve katalysatoren slechts enkele cycli kunnen werken. Een belangrijk kenmerk van de mate van metallische katalytische functionaliteit hangt af van de sterkte van adsorptie voor een verscheidenheid aan soorten op het katalysatoroppervlak. Volgens het Sabatier-principe, meer dan een eeuw geleden ontwikkeld, actieve katalysatoren moeten adsorbaten binden met een bindingssterkte die niet sterk of zwak is. Hoewel sterk geadsorbeerde soorten het katalysatoroppervlak kunnen vergiftigen, zwak gebonden reactanten desorberen gemakkelijk. In een tussenscenario de reactanten kunnen elkaar ontmoeten en reageren op de katalytische oppervlakken. Onderzoekers gebruiken momenteel computationele simulatie- en theoretische chemiemethoden om katalytisch gedrag op metaalkatalysatoren met grote nauwkeurigheid te stimuleren om latere experimenten in het laboratorium te begeleiden.
Computerinspanningen waren gericht op het screenen van verschillende metaalkatalysatoren om de "magische" bindingsenergie (BE) van chemische soorten op katalysatoroppervlakken te ontdekken om zeer actieve katalysatoren te vormen. Het in silico ontwerp van katalytisch actieve materialen, echter, moet nog gerealiseerd worden. De nadelen zijn voornamelijk te wijten aan ontwerpinspanningen die vaak de stabiliteit van katalysatoren verwaarlozen. NP-katalysatoren bezitten ook een hoge mate van heterogeniteit op hun oppervlak voor adsorptie en katalyse. Wetenschappers hadden adsorptiemodellen ontwikkeld om bindingsenergie van de adsorbaten te relateren aan oppervlaktekenmerken van NP's zoals coördinatiegetallen (CN's) om de plaatsspecifieke adsorptierespons te begrijpen. Nog, voor alle duidelijkheid, de variatie in bindingsenergie (BE) omvat ook secundaire descriptoren zoals kromming en elektronische eigenschappen van NP's.
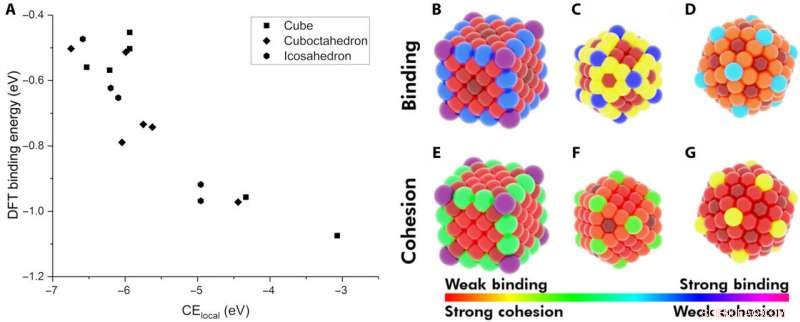
Demonstratie van lokale cohesieve energie (CElocal) als descriptor voor adsorptie-energie. (A) De BE van CO op verschillende locaties van Au NP's als een functie van CElocal:kubus van 172 atomen (rechthoeken), 147-atoom icosaëder (zeshoeken), en 147-atoom cuboctaëder (ruit). Warmtekaart van verschillende locaties op de NP's met betrekking tot hun BE van CO (B tot D) en hun CElocal (E tot G). Het kleurenschema volgt het bereik van de sterkste CO-binding tot de zwakste CElocal (violet) en van de zwakste binding tot de sterkste CElocal (rood). Krediet:wetenschappelijke vooruitgang, doi:10.1126/sciadv.aax5101
In het huidige werk, Dean et al. toegepaste dichtheidsfunctionaaltheorie (DFT) en machine learning-technieken om een eenvoudig op fysica gebaseerd model af te leiden om de variërende adsorptie-energie nauwkeurig vast te leggen. Ze schatten de variabele als een functie van de lokale adsorptieplaatsomgeving op het NP-oppervlak en het type metalen NP. Het gegeneraliseerde model kan worden toegepast op elke metalen nanostructuur om het adsorptiegedrag op de NP-katalysator en de stabiliteit van de katalysator te begrijpen; voor het screenen en ontwerpen van katalysatoren voor tal van toepassingen.
De onderzoekers veronderstelden eerst de belangrijkste factoren tussen monometallische NP's en adsorbaten. Vervolgens definieerden ze de lokale cohesieve energie (CE lokaal ) in bulkmetalen en vastgelegde CE's in NP's met behulp van een op bindingen gericht model, die elke metaal-metaalbindingsenergie optelde. Door vergelijkbare concepten toe te passen, they described the stability of binding sites and showed how chemically unsaturated sites (fewer metal-metal bonds) bound adsorbates with an increased strength. The research team focused on describing the binding capacity of a single adsorbate-metal pair. They plotted the DFT-calculated binding energy of carbon monoxide (CO) to a 172-atom gold (Au) cube and a 147-atom gold (Au) cuboctahedron or icosahedron. The team observed a strongly inverse relationship between the local cohesive energy (CE local ) and binding energy (BE) to suggest the strongest adsorption sites to be those exhibiting the weakest local cohesion.
The team further developed their model and performed ordinary least squares (OLS) regression to understand adsorption on monometallic NPs and slabs using three adsorbates [Methyl radical (CH 3 ), CO, hydroxyl radical (OH)] on three different metals (Cu, Ag—silver, Au). The metallic NPs contained different morphologies (172-atom cube, 55- and 147-atom icosahedron and 55- and 147-atom cuboctahedron). They observed that the binding affinity to the adsorbates decreased as the cohesion of the local sites increased. And as the adsorbate's chemical potential increased, they became less stable and bound a metal NP with higher tendency. The direct correlation with the metal Adsorbate (MAD) intuitively described the tendency of the metal to bind the adsorbate.

Parity plot of the model-predicted binding energy (BE) of adsorbates (OH, CO, and CH3) on various metal systems versus the DFT BE (eV). (A) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which includes the nanoparticle cohesive energy (CENP) term. (B) The model both trained and tested on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms), which does not include the CENP term. (C) The model trained on PBE DFT data for NPs (Au/Ag/Cu, 55 to 172 atoms) and tested against RPBE (revised Perdew-Burke-Ernzerhof model) DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu). (D) The model both trained and tested on RPBE DFT data for top-site adsorptions on metal surfaces (Au/Ag/Cu) from the slab dataset. Krediet:wetenschappelijke vooruitgang, doi:10.1126/sciadv.aax5101
Dean et al. tested the generalizability of the model and trained the simulation on a single metal or single morphology, although it accurately captured other metals or morphologies as well. The work provided strong evidence that the model captured the underlying physics of the binding interactions, allowing the team to extend the work from non-periodic NPs to periodic slab systems. Computationally inexpensive systems could parameterize the model to extend to larger systems, which was not thus far possible due to the computational costs involved.
Dean et al. then extended the model from monometallic NPs to bimetallic systems. For these experiments, they plotted the BE—trained on monometallic NPs, across several sites of bi-metallic, 55-atom icosahedron NPs (Cu 31 Ag 24 and Cu 22 Ag 33 ). The model very accurately captured trends in adsorption on the bimetallic Cu/Ag NPs as well. This was an interesting result since the scientists had only trained the model on monometallic systems. The results showed the generalizability of the model for both monometallic and bimetallic NPs. Echter, the team will account additional descriptors including binding site electronegativity to understand the adsorption behavior for bimetallic systems in depth.
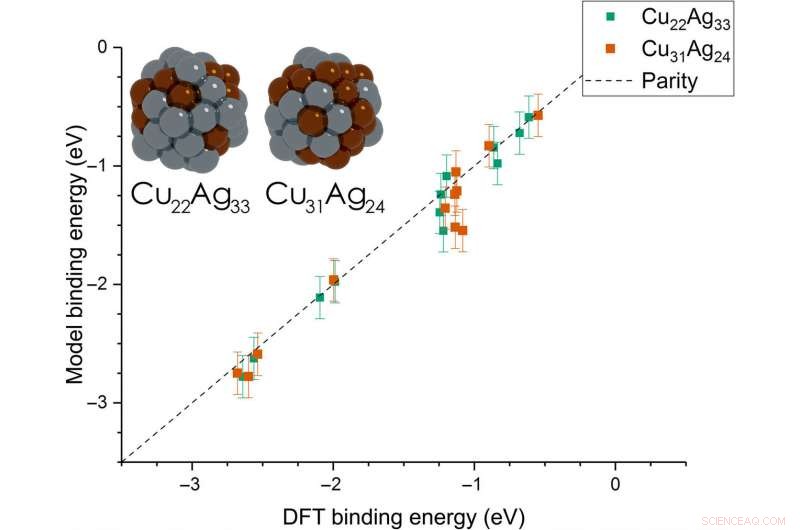
Parity plot between the presently developed model and DFT calculations on icosahedral bimetallic (Cu55−xAgx, x =24, 33) NPs. The model is trained on CH3, CO, and OH adsorbing on monometallic Ag, Cu, and Au NPs and is able to capture adsorption on bimetallic NPs. Images of the two NPs are shown as inset, with copper and silver atoms colored in brown and gray, respectievelijk. Krediet:wetenschappelijke vooruitgang, doi:10.1126/sciadv.aax5101
Although Dean et al. trained the ML (machine learning) algorithm to capture the adsorption trends of just one type of d9 metal, it could accurately predict the behavior of similar d9 metals (Cu—coper, Ag and Au). When they trained the model on a dataset of CH 3, CO and OH adsorbed to Cu, Ag and Au NPs, they could also capture general adsorption trends for similar elements in other columns of the periodic table. They then improved the complexity of the machine learning techniques to provide additional avenues to improve the model of adsorption.
Op deze manier, James Dean and his colleagues developed a simple yet powerful physics-based model to capture trends on the strength of binding interactions between different adsorbates and metal NPs using machine learning techniques. The study was the first to develop an adsorption model that accurately connected the properties of diverse metal NPs with the stability of the adsorption site. The model introduced simple descriptors to capture the adsorption on any site, relative to monometallic and bimetallic NPs. The team generalized the model to effectively stimulate a range of binding interactions, including variations on the types of metals, their composition, sites of adsorption and adsorbates.
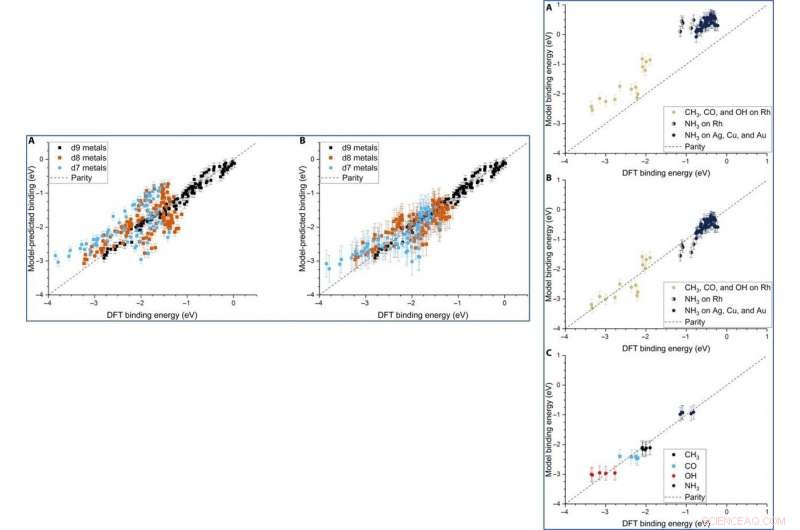
LEFT:The three-descriptor model extended to slab dataset. (A) The model trained on the slab dataset on Cu, Ag, and Au surfaces and tested against the Rh, Ir, Ni, Pd, Pt, Cu, Ag, and Au surfaces from the slab dataset. (B) The equivalent model when trained separately for each column of the d-block, still using the slab dataset. Error bars in every case are the 10-fold cross-validated RMSE of the training set. RIGHT:Extension of the model to Rh and NH3. (A) The model parameterized on our Ag, Cu, and Au NPs adsorbing CH3, CO, and OH and tested against Rh and NH3. (B) The equivalent model with empirical (constant) corrections for Rh and NH3. In the case of NH3 bound to Rh, both corrections are simultaneously applied and indicated by two-colored dots. (C) The model trained on CH3, CO, OH, and NH3 adsorbing on icosahedral/cuboctahedral Rh55. Krediet:wetenschappelijke vooruitgang, doi:10.1126/sciadv.aax5101
Although the team did not test the applicability of the model for ternary systems, the physical properties may remain relevant to accurately model multimetallic systems as well. The adsorption model can accurately describe the binding strength of a variety of molecules on any site of NPs, including alloys. The scientists expect the model to be highly applicable as a screening tool for the high throughput search of potential catalysts.
© 2019 Wetenschap X Netwerk
 Overzeese klimaatverandering kan het VK verwoesten
Overzeese klimaatverandering kan het VK verwoesten- Studie vindt dat herstel van Nachusa Grasslands werkt in de bodem, te
- Gevangen water, kooldioxide uit auto-uitlaatgassen kan helpen bij het verbouwen van voedsel
- Ellende, onzekerheid nadat Irma de idylle van Florida bereikt
- Verbetering van de waterkwaliteit van Lake Eries
Hoofdlijnen
- Studie werpt nieuw licht op hoe dieren en planten reageren op veranderingen in de omgeving
- Onderzoek bevestigt het:we worden echt dommer
- Wat zijn natuurlijke polymeren?
Enkele van de meest voorkomende voorbeelden van polymeren zijn kunststoffen en eiwitten. Hoewel plastics het resultaat zijn van het industriële proces, zijn eiwitten rijk aan aard en worden ze daarom meestal als een
- Malawi wendt zich tot Britse troepen in stropersoorlog
- Waarom hebben we enzymen nodig voor de spijsvertering?
- Evolutie:de begunstigden van massale uitsterving
- Studie kijkt naar de invloed van woonwerven op voedselwebben
- Wetenschappers ontdekken dat darmbacteriën in bijen antibioticaresistente genen naar elkaar verspreiden
- Hartmonitors op wilde narwallen onthullen alarmerende reacties op stress




 Wiskundige suggereert een schema voor het oplossen van telegraafvergelijkingen
Wiskundige suggereert een schema voor het oplossen van telegraafvergelijkingen- Astronomen vinden 72 heldere en snelle explosies
- Hoe werkt een magnetometer?
- Wat zijn enkele voorbeelden van een kustvlakte?
- Verbeterde modellering van de nucleaire structuur in francium helpt bij het zoeken naar nieuwe fysica
- Wanneer ontwikkeling en behoud botsen in de Serengeti
- Wat zijn twee manieren Wind veroorzaakt erosie?
- Video:Grondwater in kaart brengen vanuit de lucht
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Swedish | German | Dutch | Danish | Norway | Portuguese |

-
Wetenschap © https://nl.scienceaq.com
