Wetenschap
Aminozuurvingerafdrukken onthuld in nieuwe studie
Stuart Lindsay is de directeur van het Center for Single Molecule Biophysics aan het Biodesign Institute van de Arizona Arizona State University. Krediet:het Biodesign Institute aan de Arizona State University
Ongeveer drie miljard basenparen vormen het menselijk genoom - de plattegrond van het leven. In 2003, het Human Genome Project heeft de succesvolle decodering van deze code aangekondigd, een krachttoer die een stroom van inzichten blijft leveren die relevant zijn voor de menselijke gezondheid en ziekte.
Hoe dan ook, de belangrijkste actoren in vrijwel alle levensprocessen zijn de eiwitten waarvoor wordt gecodeerd door DNA-sequenties die bekend staan als genen. Voor een breed spectrum van ziekten, eiwitten kunnen veel overtuigender onthullingen opleveren dan alleen uit DNA kan worden afgeleid, als onderzoekers erin slagen de aminozuursequenties waaruit ze zijn samengesteld te ontsluiten.
Nutsvoorzieningen, Stuart Lindsay en zijn collega's van het Biodesign Institute van de Arizona State University hebben een grote stap in deze richting gezet, het aantonen van de nauwkeurige identificatie van aminozuren, door elk kort vast te pinnen in een nauwe verbinding tussen een paar flankerende elektroden en een karakteristieke reeks stroompieken te meten die door opeenvolgende aminozuurmoleculen gaan.
Door een machine learning-algoritme te gebruiken, Lindsay en zijn team waren in staat om een computer te trainen om uitbarstingen van elektrische activiteit te herkennen die de tijdelijke binding van een aminozuur in de junctie vertegenwoordigen. De ruissignalen bleken te fungeren als betrouwbare vingerafdrukken, het identificeren van aminozuren, inclusief subtiel aangepaste varianten.
Eiwitten leveren al een schat aan informatie over ziekten, waaronder kanker, diabetes en neurologische aandoeningen zoals de ziekte van Alzheimer, evenals het verschaffen van belangrijke inzichten in een ander door eiwit gemedieerd proces:veroudering.
Het nieuwe werk bevordert het vooruitzicht van klinische eiwitsequencing en de ontdekking van nieuwe biomarkers - vroege waarschuwingsbakens die ziekte signaleren. Verder, eiwitsequencing kan de behandeling van patiënten radicaal veranderen, waardoor nauwkeurige monitoring van de ziekterespons op therapieën mogelijk is, op moleculair niveau.
De onderzoeksresultaten van de groep worden gerapporteerd in de geavanceerde online editie van het tijdschrift Natuur Nanotechnologie .
Van genoom tot proteoom
Een enorme bibliotheek van eiwitten, bekend als het proteoom, staat centraal in vrijwel alle levensprocessen. Eiwitten zijn essentieel voor celgroei, differentiatie en reparatie; ze katalyseren chemische reacties en bieden bescherming tegen ziekten, tussen talloze huishoudelijke taken.
Een van de vreemdste verrassingen van het Human Genome Project is het feit dat slechts ongeveer 1,5 procent van het genoom codeert voor eiwitten. De rest van de DNA-nucleotiden vormen regulerende sequenties, niet-coderende RNA-genen, intronen, en niet-coderend DNA, (ooit spottend bestempeld als "junk-DNA"). Dit laat mensen met een karige 20-25, 000 genen, een ontnuchterende ontdekking aangezien de nederige rondworm ongeveer hetzelfde aantal heeft. Zoals professor Lindsay opmerkt, het nieuws wordt erger:"Een lelieplant heeft ongeveer een orde van grootte meer genen dan wij, " hij zegt.
Het mysterie van complexe organismen zoals mensen met een schrikbarend laag aantal genen heeft te maken met het feit dat eiwitten die worden gegenereerd op basis van de DNA-blauwdruk op een aantal manieren kunnen worden gewijzigd. In feite, wetenschappers hebben al meer dan 100 geïdentificeerd, 000 menselijke eiwitten en onderzoekers zoals Lindsay geloven dat dit slechts het topje van de ijsberg is.
Net zoals zinnen hun betekenis kunnen veranderen door veranderingen in woordvolgorde of interpunctie, eiwitten gegenereerd uit gentemplates kunnen van functie veranderen (of kunnen soms onbruikbaar worden gemaakt), vaak met ernstige gevolgen voor de menselijke gezondheid. Twee belangrijke processen die eiwitten wijzigen, staan bekend als alternatieve splicing en post-translationele modificatie. Zij zijn de aanjagers van de buitengewone eiwitvariatie die wordt waargenomen.
Alternatieve splicing vindt plaats bij het coderen van regio's van RNA, (bekend als exons) worden aan elkaar gesplitst en niet-coderende gebieden (bekend als introns) worden weggeknipt, voor translatie in eiwitten. Dit proces verloopt niet altijd netjes, met incidentele overlappingen van exons of introns die worden geïntroduceerd, het produceren van alternatief gesplitste eiwitten, waarvan de functie kan worden gewijzigd.
Post-translationele modificaties zijn markers die worden toegevoegd nadat eiwitten zijn gemaakt. Er zijn vele vormen van post-translationele modificatie, inclusief methylering en fosforylering. Sommige veranderde eiwitten vervullen vitale functies, terwijl andere afwijkend kunnen zijn en verband houden met ziekte (of neiging tot ziekte). Een aantal kankers wordt in verband gebracht met dergelijke eiwitfouten, die al worden gebruikt als diagnostische markers. Een goede identificatie van dergelijke eiwitten blijft echter een grote uitdaging in de biogeneeskunde.
Nieuwe reeksen
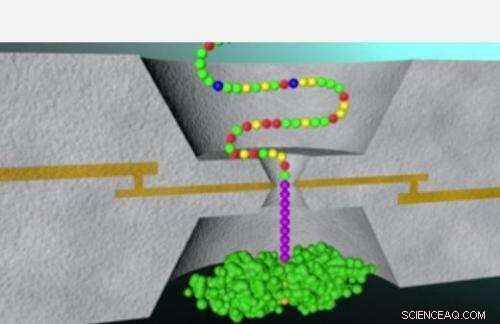
De techniek die in het huidige onderzoek wordt beschreven, werd eerder toegepast in het Lindsay-lab voor het succesvol sequencen van DNA-basen. Deze methode, bekend als herkenningstunneling, houdt in dat een peptide door een klein oogje wordt gehaald dat bekend staat als een nanoporie. Een paar metalen elektroden, gescheiden door een opening van ongeveer twee nanometer, zit aan weerszijden van de nanoporie terwijl opeenvolgende eenheden van een peptide door de kleine opening worden geregen, waarbij elke eenheid een elektrisch circuit voltooit en een uitbarsting van stroompieken uitzendt.
De onderzoeksgroep toonde aan dat nauwkeurige analyses van deze stroompieken onderzoekers in staat zouden kunnen stellen om te bepalen welke van de vier nucleotidebasen - adenine, thymine, cytosine of guanine - bevond zich tussen de elektroden in de nanoporie.
"Ongeveer 2 jaar geleden tijdens een van onze laboratoriumbijeenkomsten, er werd gesuggereerd dat misschien dezelfde technologie zou werken voor aminozuren, " zegt Lindsay. Zo begonnen de inspanningen om de aanzienlijk grotere uitdaging aan te gaan om herkenningstunneling te gebruiken om alle 20 aminozuren in eiwitten te identificeren, in tegenstelling tot slechts 4 basen die DNA bevatten.
Single-molecule sequencing van eiwitten is van enorme waarde, biedt de mogelijkheid om afnemende kleine hoeveelheden eiwitten te detecteren die mogelijk zijn aangepast door alternatieve splicing of post-translationele modificatie. Vaak, these are the very proteins of interest from the standpoint of recognizing disease states, though current technologies are inadequate to detect them.
As Lindsay notes, there is no equivalent in the protein world to polymerase chain reaction (PCR) technology, which allows minute quantities of DNA in a sample to be rapidly amplified. "We probably don't even know about most of the proteins that would be important in diagnostics. It's just a black hole to us because the concentrations are too low for current analytical techniques, " hij zegt, adding that the ability of recognition tunneling to pinpoint abnormalities on a single molecule basis "could be a complete game changer in proteomics."
The new paper describes a series of experiments in which pure samples of individual amino acids, individual molecules in mixed solution and finally, short peptide chains were successfully identified through recognition tunneling. The work sets the stage for a method to sequence individual protein molecules rapidly and cheaply (see accompanying animation).
A machine learning algorithm known as Support Vector Machine was used to train a computer to analyze the burst signals produced when amino acids formed bonds in the tunnel junction and emitted a lively noise signal as the poised electrodes passed tunneling current through each molecule. (The machine learning algorithm is the same one used by the IBM computer 'Watson' to defeat a human opponent in Jeopardy.)
Lindsay says that around 50 distinct signal burst characteristics were used in the amino acid identifications, but that most of the discriminatory power is achieved with 10 or fewer signal traits.
Opmerkelijk, recognition tunneling not only pinpointed amino acids with high reliability from single complex burst signals, but managed to distinguish a post-translationally modified protein (sarcosine) from its unmodified precursor (glycine) and also to discriminate between mirror-image molecules knows as enantiomers and so-called isobaric molecules, which differ in peptide sequence but exhibit identical masses.
Pathway to the $1000 dollar proteome?
Lindsay indicates that the new studies, which rely on innovative strategies for handling single molecules coupled with startling advances in computing power, open up horizons that were inconceivable only a short time ago. It is becoming clear that the tools that made the $1000 genome feasible are equally applicable to an eventual $1000 dollar proteome. Inderdaad, such a landmark may not be far off. "Why not?" Lindsay asks. "People think it's crazy but the technical tools are there and what will work for DNA sequencing will work for protein sequencing."
While the tunneling measurements have until now been made using a complex laboratory instrument known as a scanning tunneling microscope (STM), Lindsay and his colleagues are currently working on a solid state device capable of fast, cost-effective and clinically applicable recognition tunneling of amino acids and other analytes. Eventual application of such solid-state devices in massively parallel systems should make clinical proteomics a practical reality.
 Onderzoekers ontwikkelen materialen die een revolutie teweeg kunnen brengen in de manier waarop licht wordt gebruikt voor zonne-energie
Onderzoekers ontwikkelen materialen die een revolutie teweeg kunnen brengen in de manier waarop licht wordt gebruikt voor zonne-energie- Nieuwe membraantechnologie om waterzuivering en energieopslag te stimuleren
- Een veiligere manier voor de politie om drugsbewijs te testen
- Nieuwe tool verandert het spel voor heterogene materiaalmodellering
- Kopercoating op 3D-geprinte plastic filters voorgesteld als een pandemische strijder
- UITLEG:Waarom huisbeveiliging belangrijk is bij bosbranden
- Antarctisch schiereiland zal de komende twee decennia waarschijnlijk opwarmen
- Het IPCC oceans-rapport is een wake-up call voor beleidsmakers
- Toenemende klimaatangst vormt een aanzienlijke bedreiging voor individuen en de samenleving
- Paddenstoelachtige koralen krijgen hun genomen in kaart
Hoofdlijnen
- Onderzoekers identificeren moleculaire motor die chromosomen transformeert
- Definitie van menselijke biologie
- Cellen puilen uit om door barrières te persen
- Grafische Australische video van Japanse walvisvangst vrijgegeven
- Forensische wetenschapsprojecten voor middelbare scholieren
- Neurale opnames van wilde vleermuizen onthullen een unieke organisatie van het middenhersenengebied voor het volgen en vangen van prooien
- Exon: definitie, functie en belang in RNA Splicing
- Hebben alle cellen mitochondriën?
- Oude levensvorm ontdekt in afgelegen Tasmaanse vallei
- Nieuwe techniek om nanogestructureerde nanodraden te synthetiseren

- Siliciumalternatieven sleutel tot toekomstige computers, consumentenelektronica
- 3D nanosupergeleiders maken met DNA

- Entropiemetingen onthullen exotisch effect in grafeen met magische hoek

- Boornitride nanofilms voor bescherming tegen bacteriële en schimmelinfecties


 Onderzoekers ontwikkelen microscoop om lichtenergiestroom in fotosynthetische cellen te volgen
Onderzoekers ontwikkelen microscoop om lichtenergiestroom in fotosynthetische cellen te volgen- Naschokken rammelen Kroatische hoofdstad een dag na sterke aardbeving
- Hoe reproduceren schimmels?
- Is het mogelijk om de spullen te zien die op de maan zijn achtergelaten?
- Bevriezende regen feiten
- Reinigen zonder te schrobben en chemicaliën te gebruiken. Wetenschappers ontwikkelen zelfreinigend aluminium oppervlak
- Hoe ruimtebegraven werkt
- Onderzoekers bieden een kijkje in ecosystemen van dinosauriërs
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
