Wetenschap
Uitgebreide elektronische structuurmethoden voor materiaalontwerp
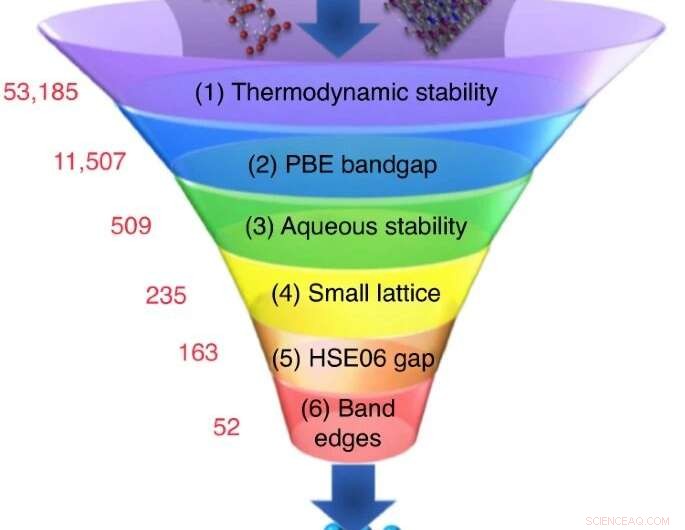
Een groot aantal kandidaat-materialen wordt gekozen uit experimentele of computationele databases, en een reeks screeningsberekeningen reduceert hun aantal tot een kleine groep kandidaten met de meest veelbelovende eigenschappen. Krediet:Nicola Marzari
Nicola Marzari, hoofd van het laboratorium Theory and Simulation of Materials bij EFPL en directeur van NCCR MARVEL, heeft zojuist een bespreking van elektronische structuurmethoden gepubliceerd als onderdeel van een speciale editie Insight on Computational Materials Design, gepubliceerd door Natuurmaterialen . Het artikel, geschreven met Andrea Ferretti van CNR-Instituto Nanoscienze en Chris Wolverton van Northwestern University, geeft een overzicht van deze methoden, bespreekt hun toepassing op de voorspelling van materiaaleigenschappen, en onderzoekt verschillende strategieën die worden gebruikt om de bredere doelen van materiaalontwerp en -ontdekking te bereiken. Vooruit kijken, de auteurs beschouwen nieuwe uitdagingen in de voorspellende nauwkeurigheid van de berekeningen, en bij het aanpakken van de real-life complexiteit van materialen en apparaten. Ze benadrukken ook het belang van de computationele infrastructuren die dergelijk onderzoek ondersteunen, en hoe de planning voor de financiering van deze en de ondersteunende loopbaanmodellen nog maar net begint te ontstaan.
In de afgelopen 20 jaar, eerste-principesimulaties zijn krachtig geworden, veelgebruikte tools in veel, verschillende gebieden van wetenschap en techniek. Van nanotechnologie tot planetaire wetenschap, van metallurgie tot kwantummaterialen, ze hebben de identificatie versneld, karakteriseren, en optimalisatie van materialen enorm. Ze hebben geleid tot verbazingwekkende voorspellingen - van ultrasnel thermisch transport tot door elektronen-fonon gemedieerde supergeleiding in hydriden tot de opkomst van platte banden in gedraaid dubbellaags grafeen - die tot opmerkelijke experimenten hebben geleid.
De huidige druk om experimenten aan te vullen met simulaties; voortgezet, snelle groei van de doorvoercapaciteit van computers; het vermogen van machine learning en kunstmatige intelligentie om de ontdekking van materialen te versnellen, evenals de belofte van ontwrichtende versnellers zoals kwantumcomputing voor exponentieel dure taken, betekent dat het duidelijk is dat deze methoden in de loop van de tijd steeds relevanter zullen worden. Het is dan ook een geschikt moment om de mogelijkheden en de beperkingen van de elektronische structuurmethoden die aan deze simulaties ten grondslag liggen, te herzien. Marzari, Ferretti en Wolverton behandelen deze taak in het artikel "Electronic-structure methods for materials design, " net gepubliceerd in Natuurmaterialen .
"Simulaties mislukken niet op spectaculaire manieren, maar kunnen subtiel verschuiven van onschatbaar naar nauwelijks goed genoeg naar gewoon nutteloos, " zeiden de auteurs in de krant. "De redenen voor het falen zijn legio, van het uitbreiden van de mogelijkheden van de methoden tot het opgeven van de complexiteit van echte materialen. Maar simulaties zijn ook onvervangbaar:ze kunnen materialen beoordelen onder omstandigheden van druk en temperatuur die zo extreem zijn dat geen enkel experiment op aarde in staat is om te repliceren, ze kunnen met steeds grotere behendigheid de enorme ruimte van materiaalfasen en composities verkennen op zoek naar die ongrijpbare materiaaldoorbraak, en ze kunnen de microscopische oorzaken en oorsprong van een macroscopische eigenschap direct identificeren. Laatste, ze delen met alle takken van computationele wetenschap een sleutelelement van onderzoek:ze kunnen reproduceerbaar en open en deelbaar worden gemaakt op manieren die geen enkele fysieke infrastructuur ooit zal zijn."
De auteurs kijken eerst naar het raamwerk van de dichtheidsfunctionele theorie (DFT) en geven een overzicht van de steeds complexere benaderingen die de nauwkeurigheid kunnen verbeteren of de reikwijdte van simulaties kunnen uitbreiden. Vervolgens bespreken ze de mogelijkheden die de computationele materiaalwetenschap heeft ontwikkeld om deze toolbox te benutten en voorspellingen te doen voor de eigenschappen van materialen onder realistische omstandigheden van steeds toenemende complexiteit. Eindelijk, ze benadrukken hoe fysica- of datagestuurde benaderingen rationele, hoge doorvoer, of kunstmatige intelligentie manieren om materialen te ontdekken, en uitleggen hoe dergelijke inspanningen het hele onderzoeksecosysteem veranderen.
Vooruit kijken, de auteurs zeggen dat het ontwikkelen van methoden die de thermodynamische stabiliteit kunnen beoordelen, synthese voorwaarden, maakbaarheid, en tolerantie van de voorspelde eigenschappen voor intrinsieke en extrinsieke defecten in nieuwe materialen zal een grote uitdaging zijn. Onderzoekers moeten mogelijk DFT-schattingen aanvullen met meer geavanceerde elektronische structuurmethoden of machine learning-algoritmen om de nauwkeurigheid te verbeteren, en computationele methoden gebruiken om realistische omstandigheden aan te pakken, zoals vibrationele entropieën, de concentratie van defecten en toegepaste elektrochemische potentialen.
Eindelijk, gezien de grotere rol die dergelijke methoden in de komende decennia waarschijnlijk zullen spelen, de auteurs merken op dat ondersteuning en planning voor de benodigde rekeninfrastructuren - veelgebruikte wetenschappelijke software, de verificatie van codes en validatie van theorieën, de verspreiding en beheer van computationele gegevens, tools en workflows, evenals de bijbehorende loopbaanmodellen die deze met zich meebrengen en vereisen, beginnen nog maar net te ontstaan.
 Onderzoekers creëren sterke, snel, waterdichte lijm
Onderzoekers creëren sterke, snel, waterdichte lijm- Wetenschappers maken belangrijke doorbraak op weg naar nieuw superbug-dodend antibioticum teixobactine
- Technische defecten in kristallijn materiaal verhogen de elektrische prestaties
- Nieuwe biofarmaceutische kwaliteitscontrolemethode bij testen
- Hoe maak je een Atoomreplica van Uranium voor School
- Trans-Himalaya land van Upper Mustang in Nepal kan te maken krijgen met ernstige voedselonzekerheid
- Hoe zijn voedselketens en voedselwebsites hetzelfde en verschillend?
- Vleermuispoep kan onderzoekers aanwijzingen geven over historische veranderingen in het klimaat, vegetatie
- Niet genoeg hazelnoten? Ons toekomstige klimaat wijst naar Australië voor nieuwe teelt
- Tropische bergrivieren zijn waar de magie gebeurt
Hoofdlijnen
- Soorten menselijke schedelvormen
- Hoe antibioticagebruik bij dieren bijdraagt aan antibioticaresistentie
- mensen, in tegenstelling tot apen, verander de concurrentiesituatie in een coöperatieve
- Een veranderend klimaat, wijn wisselen
- Plantaanpassingen: woestijn, tropisch regenwoud, toendra
- Maken regenachtige dagen je echt down?
- Veel meer bacteriën hebben elektrisch geleidende filamenten
- De rol van microben in de industrie
- Vliegen ontwikkelen springen zonder poten
- Wat is de CO2-voetafdruk van een plastic fles?

- Ontdekken hoe T-cellen het SARS-COV-2 virus spike-eiwit herkennen
- Nobelprijswinnende techniek zoals Google Earth voor moleculen

- Onderzoekers krijgen een glimp op nanoschaal van spleet- en putcorrosie terwijl het gebeurt

- As uit elektriciteitscentrales verwerkt tot reagentia voor waterzuivering


 Topologische defecten kunnen de sleutel zijn tot toekomstige nano-elektronica
Topologische defecten kunnen de sleutel zijn tot toekomstige nano-elektronica- Tijd die ouders met kinderen doorbrengen, de sleutel tot academisch succes
- Ultrahogedruklaserexperimenten werpen licht op kernen van superaarde
- Methoden voor het volgen van microbiële fecale vervuiling in water
- Onderzoekers bieden overzicht van samengestelde metaalschuimen en mogelijke toepassingen
- Dodental stijgt tot 11 in Spanje nu storm Gloria wegebt
- Een gewoon plastic omzetten in hoogwaardige moleculen
- Hoe leid je cultureel competente muziekleraren op?
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
