Wetenschap
Moleculaire dynamiek, machine learning maakt hypervoorspellende computermodellen
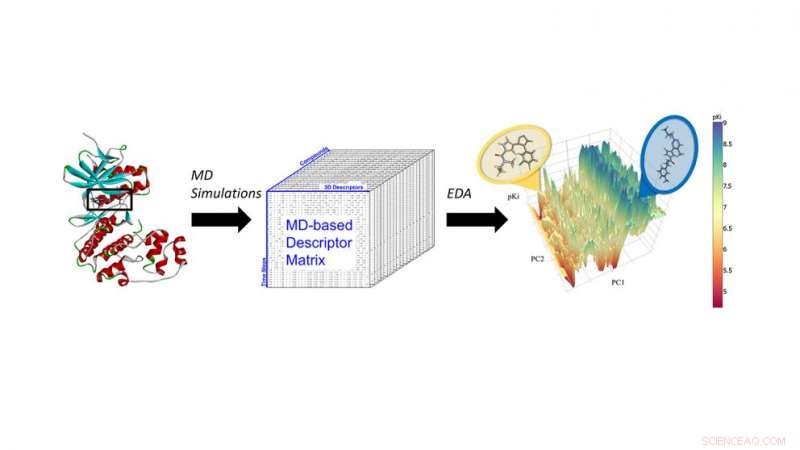
Moleculaire dynamica (MD) simulaties van ERK2-remmers om MD-descriptoren te extraheren voor de volgende generatie cheminformatica-analyse en machine learning. Krediet:North Carolina State University
Onderzoekers van de North Carolina State University hebben aangetoond dat moleculaire dynamica-simulaties en machine learning-technieken kunnen worden geïntegreerd om nauwkeurigere computervoorspellingsmodellen te creëren. Deze "hypervoorspellende" modellen kunnen worden gebruikt om snel te voorspellen welke nieuwe chemische verbindingen veelbelovende kandidaat-geneesmiddelen kunnen zijn.
De ontwikkeling van geneesmiddelen is een kostbaar en tijdrovend proces. Om het aantal chemische verbindingen dat potentiële kandidaat-geneesmiddelen zouden kunnen zijn, te beperken, wetenschappers gebruiken computermodellen die kunnen voorspellen hoe een bepaalde chemische verbinding kan interageren met een biologisch doelwit van belang - bijvoorbeeld, een belangrijk eiwit dat mogelijk betrokken is bij een ziekteproces. traditioneel, dit gebeurt via kwantitatieve structuur-activiteitsrelatie (QSAR) -modellering en moleculaire docking, die afhankelijk zijn van 2- en 3D-informatie over die chemicaliën.
Denis Fourches, assistent-professor computationele chemie, wilde de nauwkeurigheid van deze QSAR-modellen verbeteren. "Als je een set van 30 miljoen verbindingen screent, je hebt niet per se een zeer hoge betrouwbaarheid nodig met je model - je krijgt gewoon een idee van de top 5 of 10 procent van die virtuele bibliotheek. Maar als je een veld van 200 analogen probeert te verkleinen tot 10, wat vaker het geval is bij de ontwikkeling van geneesmiddelen, uw modelleringstechniek moet uiterst nauwkeurig zijn. De huidige technieken zijn absoluut niet betrouwbaar genoeg."
Fourches en Jeremy Ash, een afgestudeerde student bio-informatica, besloot om de resultaten van moleculaire dynamica-berekeningen - all-atom-simulaties van hoe een bepaalde verbinding in de bindingszak van een eiwit beweegt - op te nemen in voorspellingsmodellen op basis van machine learning.
"De meeste modellen gebruiken alleen de tweedimensionale structuren van moleculen, " zegt Fourches. "Maar in werkelijkheid, chemicaliën zijn complexe driedimensionale objecten die bewegen, trillen en hebben dynamische intermoleculaire interacties met het eiwit als het eenmaal op zijn bindingsplaats is gedokt. Dat kun je niet zien als je alleen naar de 2D- of 3D-structuur van een bepaald molecuul kijkt."
In een proof-of-concept-onderzoek Fourches en Ash keken naar het ERK2-kinase - een enzym geassocieerd met verschillende soorten kanker - en een groep van 87 bekende ERK2-remmers, variërend van zeer actief tot inactief. Ze voerden onafhankelijke moleculaire dynamica (MD) simulaties uit voor elk van die 87 verbindingen en berekenden kritische informatie over de flexibiliteit van elke verbinding eenmaal in de ERK2-pocket. Vervolgens analyseerden ze de MD-descriptoren met behulp van cheminformatics-technieken en machine learning. De MD-descriptoren waren in staat om actieve ERK2-remmers nauwkeurig te onderscheiden van zwak actieve en inactieven, wat niet het geval was wanneer de modellen alleen 2-D en 3-D structurele informatie gebruikten.
"We hadden al gegevens over deze 87 moleculen en hun activiteit bij ERK2, " zegt Fourches. "Dus we hebben getest om te zien of ons model in staat was om op betrouwbare wijze de meest actieve verbindingen te vinden. Inderdaad, het maakte nauwkeurig onderscheid tussen sterke en zwakke ERK2-remmers, en omdat MD-descriptoren de interacties codeerden die die verbindingen creëren in de zak van ERK2, het gaf ons ook meer inzicht waarom de sterke remmers goed werkten.
"Voordat we met geavanceerde computertechnologie dit soort gegevens konden simuleren, het zou ons zes maanden hebben gekost om één enkel molecuul in de zak van ERK2 te simuleren. Dankzij GPU-versnelling, nu duurt het maar drie uur. Dat is een gamechanger. Ik heb goede hoop dat het opnemen van gegevens die zijn geëxtraheerd uit moleculaire dynamica in QSAR-modellen een nieuwe generatie hypervoorspellende modellen mogelijk zal maken die nieuwe, effectieve medicijnen nog sneller op de markt. Het is kunstmatige intelligentie die voor ons werkt om de medicijnen van morgen te ontdekken."
 Brandstofaërosolen die de vervuiling van het milieu verminderen
Brandstofaërosolen die de vervuiling van het milieu verminderen- Een materiaalgeheugen gebruiken om unieke fysieke eigenschappen te coderen
- Filmtechnologie inspireert draagbare vloeistofeenheid die energie wil oogsten
- Nieuw enzym breekt afval af voor goedkopere biobrandstoffen, bioproducten
- Nieuwe synthetische methode voor waterstabiele perovskieten
- Onderzoek naar explosief vulkanisme tijdens ijstijd biedt lessen voor de stijgende CO2
- Duistere meren overtreffen nu duidelijk, blauwe meren in de VS
- Onderzoekers ontwikkelen systeem voor vroegtijdige waarschuwing voor watervervuiling met behulp van kleine watervlooien
- Frankrijk herziet temperatuurrecord juni tot 46 graden
- Soorten fossielen en hoe ze zijn gevormd
Hoofdlijnen
- Plankton is de kleinste onbezongen held op aarde
- Commensalisme:ik profiteer,
- Onderzoekers observeren enzymen die cellulose afbreken om de productie van biobrandstoffen te ondersteunen
- Een JELL-O-model van een diercel maken
- Verschillende soorten brood Mold
- Genetische barcodes worden gebruikt om cruciale populaties in een ecosysteem van koraalriffen te kwantificeren
- Een eenvoudig diercelmodel maken
- Voeding die de productiviteit in de intensieve veehouderij verhoogt
- 5 manieren waarop je hersenen je emoties beïnvloeden
- Onderzoekers ontwikkelen selectieve elektrokatalysatoren om de prestaties van directe methanol-brandstofcellen te verbeteren
- Fluorescerend molecuul verraadt de afbraak van polymeermaterialen

- Hybride materiaal kan in verschillende toepassingen beter presteren dan grafeen

- Een nieuw paradigma van materiaalidentificatie op basis van grafentheorie
- Betere katalysatoren voor een duurzame bio-economie


 Wetenschappers voorspellen nieuwe superharde materialen
Wetenschappers voorspellen nieuwe superharde materialen- Australische telescoop vindt geen tekenen van buitenaardse technologie in 10 miljoen sterrenstelsels
- Voor identieke kwantumkanalen, orde zaken
- Ruimtestralingsdetector kan nep-meesterwerken helpen herkennen
- Hoe is biologisch rundvlees verheven?
- NASA's Terra Satellite vindt krachtige stormen in orkaan Hector
- Californië negeert plannen voor hogesnelheidslijn LA-SF
- NASA's Lucy-missie bevestigt ontdekking van Eurybates-satelliet
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | German | Dutch | Danish | Norway | Swedish |

-
Wetenschap © https://nl.scienceaq.com
