Wetenschap
Nieuw kwantumalgoritme lost kritiek kwantumchemieprobleem op door aanpassing langs een geometrisch pad
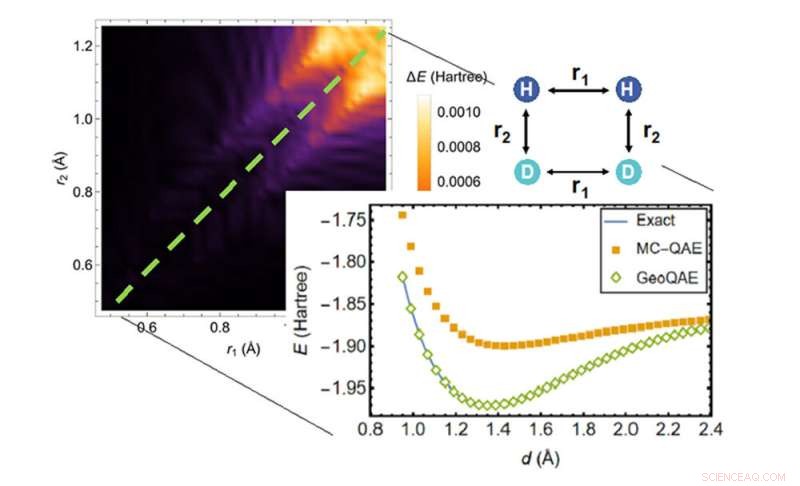
Bij het berekenen van het potentiële energieoppervlak van de chemische reactie van H2;+ D2 → 2HD, presteert het nieuwe algoritme (groene ruiten) beter dan het vorige algoritme (oranje vierkanten) bij het vinden van de meest nauwkeurige oplossing (blauwe lijn). Krediet:Brookhaven National Laboratory
Een team van onderzoekers van het Brookhaven National Laboratory van het Amerikaanse Department of Energy (DOE) en de Stony Brook University hebben een nieuw kwantumalgoritme bedacht om de laagste energieën van moleculen bij specifieke configuraties te berekenen tijdens chemische reacties, ook wanneer hun chemische bindingen worden verbroken. Zoals beschreven in Physical Review Research , vergeleken met vergelijkbare bestaande algoritmen, inclusief de vorige methode van het team, zal het nieuwe algoritme het vermogen van wetenschappers om nauwkeurig en betrouwbaar het potentiële energieoppervlak in reagerende moleculen te berekenen, aanzienlijk verbeteren.
Voor dit werk werkte Deyu Lu, een centrum voor functionele nanomaterialen (CFN) fysicus bij Brookhaven Lab, samen met Tzu-Chieh Wei, een universitair hoofddocent gespecialiseerd in kwantuminformatiewetenschap aan de C.N. Yang Institute for Theoretical Physics aan de Stony Brook University, Qin Wu, een theoreticus bij CFN, en Hongye Yu, een Ph.D. student aan Stony Brook.
"Het begrijpen van de kwantummechanica van een molecuul, hoe het zich op atomair niveau gedraagt, kan belangrijk inzicht verschaffen in zijn chemische eigenschappen, zoals zijn stabiliteit en reactiviteit", zei Lu.
Een specifieke eigenschap die een uitdaging was om te bepalen, is de grondtoestand van een molecuul:het punt waar de totale elektronische energie van het molecuul (inclusief kinetische en potentiële energie) het laagst is en niets buiten dat "moleculaire systeem" de molecuul opwindt of oplaadt. elektronen. Wanneer de atomaire structuur van een chemisch systeem complexer wordt, zoals in een groot molecuul, kunnen veel meer elektronen interageren. Die interacties maken het berekenen van de grondtoestand van complexe moleculen uiterst moeilijk.
Het nieuwe kwantumalgoritme verbetert het vorige algoritme om dit probleem op een creatieve manier aan te pakken. Het maakt gebruik van een vloeiende, geometrische vervorming die wordt veroorzaakt door continu variërende bindingslengtes of bindingshoeken in de structuur van het molecuul. Met deze aanpak zeggen de wetenschappers dat ze de grondtoestand van moleculen zeer nauwkeurig kunnen berekenen, zelfs als chemische bindingen breken en hervormen tijdens chemische reacties.
De basis leggen
"Als je alleen vertrouwt op traditionele computermethoden, bevat dit grondtoestandsprobleem te veel variabelen om op te lossen, zelfs op de krachtigste supercomputers", zegt Lu.
Je kunt een algoritme zien als een reeks stappen om een bepaald probleem op te lossen. Klassieke computers kunnen complexe algoritmen uitvoeren, maar naarmate ze groter en meer betrokken worden, kunnen ze te moeilijk of tijdrovend worden voor klassieke computers om op een haalbare manier op te lossen. Kwantumcomputers kunnen het proces versnellen door gebruik te maken van de regels van de kwantummechanica.
Bij klassieke informatica worden gegevens opgeslagen in bits met een waarde van 1 of 0. Een kwantumbit, ook wel een qubit genoemd, kan een waarde hebben die groter is dan 0 of 1, het kan zelfs een waarde hebben van 0 en 1, in een zogenaamde kwantumsuperpositie. In principe kunnen deze meer "flexibele" qubits een grotere hoeveelheid informatie opslaan dan klassieke bits. Als wetenschappers manieren kunnen vinden om de informatiedragende capaciteit van qubits te benutten, kan de rekenkracht exponentieel toenemen met elke extra qubit.
Qubits zijn echter behoorlijk kwetsbaar. Ze kunnen vaak kapot gaan wanneer informatie wordt geëxtraheerd. Wanneer een kwantumapparaat interageert met de omgeving, kan het ruis of interferentie genereren die de kwantumtoestand vernietigt. Temperatuurveranderingen, trillingen, elektromagnetische interferentie en zelfs materiaaldefecten kunnen er ook toe leiden dat qubits informatie verliezen.
Om deze valkuilen te compenseren, hebben wetenschappers een hybride oplossing ontwikkeld die gebruik maakt van zowel klassieke computeralgoritmen, die stabieler en praktischer zijn.
Lu en Wei begonnen in 2019 met onderzoek naar hybride klassieke en kwantumcomputerbenaderingen. Deze jaarlijkse subsidie bevordert de samenwerking tussen Brookhaven National Laboratory en Stony Brook University door gezamenlijke onderzoeksinitiatieven te financieren die aansluiten bij de missies van beide instellingen. Met dit eerste werk concentreerden Lu en Wei zich eerst op het oplossen van het grondtoestandsprobleem door de meest "dure" klassieke algoritmen te vervangen - degenen die veel complexer waren en aanzienlijk meer stappen (en meer rekentijd) nodig hadden om te voltooien - door kwantumalgoritmen .
Verbindingen strekken, nieuwe wegen creëren
De onderzoekers merken op dat bestaande kwantumalgoritmen allemaal nadelen hebben voor het oplossen van het grondtoestandsprobleem, waaronder het probleem dat Wei en Yu in 2019 ontwikkelden. dimensies - die algoritmen kunnen onbetrouwbaar worden wanneer de chemische bindingen op grote atomaire afstanden worden verbroken. Bindingsvorming en dissociatie spelen een rol in veel toepassingen, zoals het voorspellen hoeveel energie het kost om een chemische reactie op gang te brengen, dus wetenschappers hadden een manier nodig om dit probleem aan te pakken terwijl moleculen reageren. Ze hadden nieuwe kwantumalgoritmen nodig die het verbreken van bindingen kunnen beschrijven.
Voor deze nieuwe versie van het algoritme werkte het team samen met het door Brookhaven-Lab geleide Co-design Center for Quantum Advantage (C2QA), dat in 2020 werd opgericht. Wei draagt bij aan de softwareontwikkeling van het centrum, dat gespecialiseerd is in kwantumalgoritmen. Het nieuwe algoritme van het team gebruikt een adiabatische benadering - een die geleidelijke veranderingen aanbrengt - maar met enkele aanpassingen die ervoor zorgen dat het betrouwbaar blijft wanneer chemische bindingen worden verbroken.
"Een adiabatisch proces werkt door de omstandigheden van een kwantummechanisch systeem geleidelijk aan aan te passen", legt Lu uit. "In zekere zin bereik je in zeer kleine stappen een oplossing. Je evolueert het systeem van een eenvoudig, oplosbaar model naar het uiteindelijke doel, meestal een moeilijker model. Naast de grondtoestand echter, een veel-elektronisch systeem heeft veel aangeslagen toestanden bij hogere energieën. Die aangeslagen toestanden kunnen een uitdaging vormen bij het gebruik van deze methode om de grondtoestand te berekenen."
Wei vergeleek een adiabatisch algoritme met rijden langs een snelweg, "als je van de ene stad naar de andere reist, zijn er verschillende paden om er te komen, maar je wilt de veiligste en meest efficiënte vinden."
In het geval van kwantumchemie is de sleutel om een voldoende grote "energiekloof" te vinden tussen de grondtoestand en de aangeslagen toestanden waar geen elektronentoestanden bestaan. Met een tussenruimte die groot genoeg is, zullen de voertuigen in de metafoor van de snelweg "rijstroken niet oversteken", zodat hun paden nauwkeurig kunnen worden getraceerd.
"Een groot gat betekent dat je sneller kunt gaan, dus in zekere zin probeer je een minder drukke snelweg te vinden om sneller te rijden zonder iets te raken," zei Wei.
"Met deze algoritmen is de ingang van het pad een goed gedefinieerde, eenvoudige oplossing van klassiek computergebruik," merkte Wei op. "We weten ook waar de uitgang is - de grondtoestand van het molecuul - en we probeerden een manier te vinden om het op de meest natuurlijke manier met de ingang te verbinden, een rechte lijn.
"Dat deden we in onze eerste paper, maar de rechte lijn had wegversperringen veroorzaakt door het sluiten van de energiekloof en het kruisen van paden. Nu hebben we een betere oplossing."
Toen de wetenschappers het algoritme testten, toonden ze aan dat zelfs met eindige veranderingen in de bindingslengte, de verbeterde versie nog steeds nauwkeurig presteerde voor de grondtoestand.
"We zijn buiten onze comfortzone gegaan, want chemie is niet onze focus", zei Wei. "Maar het was goed om zo'n toepassing te vinden en een dergelijke samenwerking met CFN te bevorderen. Het is belangrijk om verschillende perspectieven in onderzoek te hebben."
Hij merkte de geaccumuleerde inspanning van veel mensen op. "In het grote plan denk ik dat we een kleine bijdrage leveren, maar dit zou een basis kunnen zijn voor ander werk op deze gebieden", zei hij. "Dit onderzoek is niet alleen fundamenteel, maar een geweldige illustratie van hoe verschillende instellingen en faciliteiten kunnen samenkomen om hun expertisegebieden te benutten." + Verder verkennen
Naar een kwantumcomputer die moleculaire energie berekent

Hoofdlijnen
- Voordelen van biosolids verspreid over tientallen jaren van onderzoek
- Veranderingen in mariene ecosystemen blijven onopgemerkt
- Wat is de rol van glucose in het lichaam?
- Hoe zou zout Gist beïnvloeden?
- Bladeren, grazen, paren:voedsel en gezelschap helpen dieren in gevangenschap
- Gedachtenexperiment:wat is ons transhumane pad voorbij de aarde?
- Anatomie en fysiologie van een synapsenstructuur
- Wat is een allel?
- Amylase Activity in the Stomach
- Alternatief materiaal voor supergeleidende radiofrequentieholte

- Gefrustreerde magneten koken op zoek naar supergeleiding
- Natuurkundigen leren de regels van magnetische toestanden in nieuw gepubliceerd onderzoek

- Voordelen en nadelen van hydraulische systemen

- Eerste onderwatertapijtmantel gerealiseerd met metamateriaal

 Gas bereikt jonge sterren langs magnetische veldlijnen
Gas bereikt jonge sterren langs magnetische veldlijnen- De beste zeoliet bouwen
- Typen radioactief verval: Alpha, Beta, Gamma
- Elektrisch circuit gemaakt van gel kan zichzelf repareren
- 3D-printen van plastic onderdelen verbeteren
- Een combinatie van plantaardige deeltjes en water vormt een eco-lijm
- Een grote kijk krijgen op kleine deeltjes
- LSU-onderzoekers werken verder aan buigbaar beton, voeg suikerrietbijproducten toe aan de mix
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
