Wetenschap
Hoe de energie van een molecuul te meten met behulp van een kwantumcomputer?
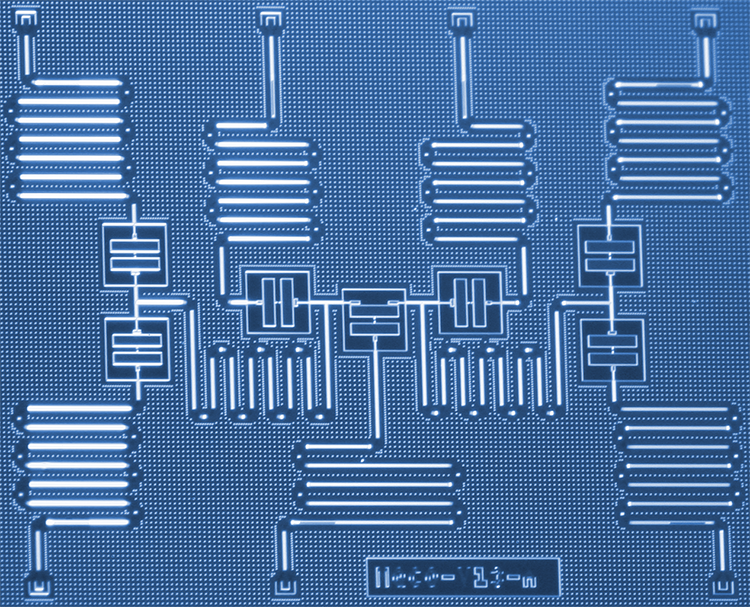
IBM-wetenschappers hebben een nieuwe benadering ontwikkeld om moleculen op een kwantumcomputer te simuleren die op een dag een revolutie teweeg kunnen brengen in de chemie en materiaalwetenschap. De wetenschappers gebruikten met succes zes qubits op een speciaal gebouwde kwantumprocessor van zeven qubits om het moleculaire structuurprobleem voor berylliumhydride (BeH2) aan te pakken - het grootste molecuul dat tot nu toe op een kwantumcomputer is gesimuleerd. De resultaten demonstreren een pad van verkenning voor kwantumsystemen op korte termijn om ons begrip van complexe chemische reacties die tot praktische toepassingen kunnen leiden, te verbeteren. Krediet:Kandala et al.; Natuur
Het simuleren van moleculen op kwantumcomputers is nu veel eenvoudiger geworden met de supergeleidende kwantumhardware van IBM. In een recent onderzoeksartikel gepubliceerd in Natuur , Hardware-efficiënte Variational Quantum Eigensolver voor kleine moleculen en kwantummagneten, we implementeren een nieuw kwantumalgoritme dat in staat is om de laagste energietoestand van kleine moleculen efficiënt te berekenen. Door de elektronische structuur van moleculaire orbitalen in kaart te brengen op een subset van onze speciaal gebouwde kwantumprocessor van zeven qubits, we bestudeerden moleculen die voorheen onontgonnen waren met kwantumcomputers, waaronder lithiumhydride (LiH) en berylliumhydride (BeH2). De specifieke codering van orbitalen naar qubits die in dit werk zijn bestudeerd, kan worden gebruikt om simulaties van nog grotere moleculen te vereenvoudigen en we verwachten de mogelijkheid om dergelijke grotere simulaties in de toekomst te verkennen, wanneer de kwantumcomputerkracht (of "kwantumvolume") van IBM Q-systemen is toegenomen.
Hoewel BeH2 het grootste molecuul is dat ooit door een kwantumcomputer is gesimuleerd, het weloverwogen model van het molecuul zelf is nog steeds eenvoudig genoeg voor klassieke computers om precies te simuleren. Dit maakte het een testcase om de grenzen te verleggen van wat onze zeven qubit-processor kon bereiken, verder ons begrip van de vereisten om de nauwkeurigheid van onze kwantumsimulaties te verbeteren, en de fundamentele elementen te leggen die nodig zijn voor het onderzoeken van dergelijke moleculaire energiestudies.
De beste simulaties van moleculen van tegenwoordig worden uitgevoerd op klassieke computers die complexe benaderingsmethoden gebruiken om de laagste energie van een moleculaire Hamiltoniaan te schatten. Een "Hamiltoniaan" is een kwantummechanische energie-operator die de interacties beschrijft tussen alle elektronenorbitalen en kernen van de samenstellende atomen. De "laagste energie" toestand van de moleculaire Hamiltoniaan dicteert de structuur van het molecuul en hoe het zal interageren met andere moleculen. Dergelijke informatie is van cruciaal belang voor chemici om nieuwe moleculen te ontwerpen, reacties, en chemische processen voor industriële toepassingen.
Qubit:Orbitaal
Hoewel onze kwantumprocessor van zeven qubits niet volledig foutgecorrigeerd en fouttolerant is, de coherentietijden van de afzonderlijke qubits duren ongeveer 50 µs. Het is dus echt belangrijk om een zeer efficiënt kwantumalgoritme te gebruiken om het meeste uit onze kostbare kwantumcoherentie te halen en moleculaire structuren te proberen te begrijpen. Het algoritme moet efficiënt zijn in termen van het aantal gebruikte qubits en het aantal uitgevoerde kwantumbewerkingen.

Toepassing op de kwantumchemie. a–c, Experimentele resultaten (zwart gevulde cirkels), exacte energie-oppervlakken (stippellijnen) en dichtheidsgrafieken (arcering; zie kleurenschalen) van uitkomsten van numerieke simulaties, voor verschillende interatomaire afstanden voor H2 (a), LiH (b) en BeH2 (c). De gepresenteerde experimentele en numerieke resultaten zijn voor circuits met diepte d = 1. De foutbalken op de experimentele gegevens zijn kleiner dan de grootte van de markeringen. De dichtheidsgrafieken worden verkregen uit 100 numerieke uitkomsten op elke interatomaire afstand. De bovenste inzetstukken in elk paneel markeren de qubits die voor het experiment zijn gebruikt en de kruisresonantiepoorten (pijlen, gelabeld CRc-t; waarbij 'c' de controle-qubit aangeeft en 't' de doel-qubit) die UENT vormen. De onderste inzetstukken zijn representaties van de moleculaire geometrie (niet op schaal). Voor alle drie de moleculen geldt de afwijking van de experimentele resultaten van de exacte curven wordt goed verklaard door de stochastische simulaties. Krediet:Kandala et al.; Natuur
Ons schema contrasteert met eerder bestudeerde kwantumsimulatie-algoritmen, die zich richten op het aanpassen van klassieke moleculaire simulatieschema's aan kwantumhardware - en daarbij niet effectief rekening houden met de beperkte overheadkosten van de huidige realistische kwantumapparaten.
Dus in plaats van klassieke computermethoden op kwantumhardware te dwingen, we hebben de aanpak omgekeerd en gevraagd:hoe kunnen we de maximale kwantumcomputerkracht uit onze zeven qubit-processor halen?
Ons antwoord hierop combineert een aantal hardware-efficiënte technieken om het probleem aan te pakken:
- Eerst, de fermionische Hamiltoniaan van een molecuul wordt omgezet in een qubit Hamiltoniaan, met een nieuwe efficiënte mapping die het aantal qubits dat nodig is in de simulatie vermindert.
- Een hardware-efficiënt kwantumcircuit dat gebruik maakt van de natuurlijk beschikbare poortbewerkingen in de kwantumprocessor, wordt gebruikt om proefgrondtoestanden van de Hamiltoniaan voor te bereiden.
- De kwantumprocessor wordt naar de proefgrondtoestand gestuurd, en metingen worden uitgevoerd waarmee we de energie van de voorbereide proeftoestand kunnen evalueren.
- De gemeten energiewaarden worden toegevoerd aan een klassieke optimalisatieroutine die het volgende kwantumcircuit genereert om de kwantumprocessor naartoe te sturen, om de energie verder te verminderen.
- Iteraties worden uitgevoerd totdat de laagste energie met de gewenste nauwkeurigheid is verkregen.
Met toekomstige kwantumprocessors, dat meer kwantumvolume zal hebben, we zullen in staat zijn om de kracht van deze benadering van kwantumsimulatie te onderzoeken voor steeds complexere moleculen die de klassieke computermogelijkheden te boven gaan. Het vermogen om chemische reacties nauwkeurig te simuleren, is bevorderlijk voor de inspanningen om nieuwe medicijnen te ontdekken, meststoffen, zelfs nieuwe duurzame energiebronnen.
De experimenten die we in onze paper beschrijven, zijn niet uitgevoerd op onze momenteel openbaar beschikbare vijf qubit- en 16 qubit-processors in de cloud. Maar ontwikkelaars en gebruikers van de IBM Q-ervaring hebben nu toegang tot kwantumchemie Jupyter-notebooks op de QISKit github-repo. Op het vijf qubit-systeem, gebruikers kunnen grondtoestand-energiesimulatie verkennen voor de kleine moleculen waterstof en LiH. Notebooks voor grotere moleculen zijn beschikbaar voor degenen met bètatoegang tot de verbeterde 16-qubit-processor.
 De effecten van water tijdens een titratie-experiment
De effecten van water tijdens een titratie-experiment- 3D-geprint apparaat vindt naald in hooiberg kankercellen door hooi te verwijderen
- Waarom scheren zelfs de scherpste scheermessen dof maakt
- Een manier om de absolute stereochemie van kleine, organische moleculen
- Wetenschappers onderzoeken toekomstig gebruik van op bacteriën gebaseerde actieve stoffen
- Vulkaanuitbarstingen veroorzaakten ooit massale uitstervingen in de oceanen - zou klimaatverandering hetzelfde kunnen doen?
- Hoe helpt het recyclen van aluminium blikjes het milieu?
- Bevestiging van een poollichtverschijnsel
- De effecten van zure regen op monumenten
- Californië registreert droogste jaar in eeuw
Hoofdlijnen
- Hoe worden bacteriën resistent tegen antibiotica?
- Vogelweervrienden:studenten uit Arkansas printen 3-D eendenpoot
- Nucleic Acid Facts
- Hoe maak ik een 7e-graads schoolproject van een virusmodel?
- Meer dan een getallenspel:nieuwe techniek meet microbiële gemeenschappen op biomassa
- Hoe leeuwenbekken hun kleur behouden:bewegwijzeringstruc onthult evolutionair mechanisme
- Het belang van samengestelde microscopen
- Wat maakt bodem, bodem? Onderzoekers vinden verborgen aanwijzingen in DNA
- Wat zijn de niveaus van organisatie in de biologie?





 Ontwerpfouten in Universal Credit voor koppels onthuld naarmate claims toenemen
Ontwerpfouten in Universal Credit voor koppels onthuld naarmate claims toenemen- Waarom de oproepen voor de defunding van de politie?
- Wat is het verschil tussen ethanol en alcohol?
- Oh, mijn sterren en zeshoeken! DNA-code vormt gouden nanodeeltjes
- Beperkende factoren in een toendra
- EU vertelt techreuzen om nepnieuws tegen het einde van het jaar aan te pakken
- Bestrijding van kunstmatige trilhaartjes met magnetische velden en licht
- Hoe Polyethyleen te maken
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
