Wetenschap
Softwareraamwerk dat is ontworpen om de ontdekking van geneesmiddelen te versnellen, wint de IEEE International Scalable Computing Challenge

Shantenu Jha, voorzitter van Brookhaven Lab's Center for Data-Driven Discovery, en zijn team van Rutgers University en University College London ontwierpen een softwareraamwerk om nauwkeurig en snel te berekenen hoe sterk kandidaat-geneesmiddelen binden aan hun doeleiwitten. Het raamwerk is gericht op het oplossen van het echte probleem van medicijnontwerp - momenteel een langdurig en duur proces - en zou een impact kunnen hebben op gepersonaliseerde geneeskunde. Krediet:Brookhaven National Laboratory
Oplossingen voor veel echte wetenschappelijke en technische problemen - van het verbeteren van weermodellen en het ontwerpen van nieuwe energiematerialen tot het begrijpen hoe het universum is gevormd - vereisen toepassingen die kunnen worden geschaald tot een zeer groot formaat en hoge prestaties. Elk jaar, via de International Scalable Computing Challenge (SCALE), het Institute of Electrical and Electronics Engineers (IEEE) erkent een project dat de ontwikkeling van applicaties en ondersteunende infrastructuur bevordert om grootschalige, high-performance computing die nodig is om dergelijke problemen op te lossen.
De winnaar van dit jaar, "Afweging mogelijk maken tussen nauwkeurigheid en rekenkosten:adaptieve algoritmen om de tijd tot klinisch inzicht te verkorten, " is het resultaat van een samenwerking tussen scheikundigen en computer- en computerwetenschappers van het Brookhaven National Laboratory van het Amerikaanse Department of Energy (DOE), Rutgers Universiteit, en University College Londen. De teamleden werden gehuldigd tijdens het 18e IEEE/Association for Computing Machinery (ACM) International Symposium on Cluster, Cloud en Grid Computing gehouden in Washington, gelijkstroom, van 1 tot 4 mei.
"We hebben een numerieke berekeningsmethode ontwikkeld voor het nauwkeurig en snel evalueren van de werkzaamheid van verschillende kandidaat-geneesmiddelen, " zei teamlid Shantenu Jha, voorzitter van het Center for Data-Driven Discovery, onderdeel van het Computational Science Initiative van Brookhaven Lab. "Hoewel we deze methode nog niet hebben toegepast om een nieuw medicijn te ontwerpen, we hebben aangetoond dat het zou kunnen werken op de grote schaal die betrokken is bij het ontdekkingsproces van geneesmiddelen."
Het ontdekken van medicijnen is als het ontwerpen van een sleutel die in een slot past. Om een medicijn effectief te laten zijn bij de behandeling van een bepaalde ziekte, het moet stevig binden aan een molecuul - meestal een eiwit - dat met die ziekte is geassocieerd. Alleen dan kan het medicijn de functie van het doelmolecuul activeren of remmen. Onderzoekers kunnen 10 screenen, 000 of meer moleculaire verbindingen voordat u er een vindt met de gewenste biologische activiteit. Maar deze "lood" verbindingen missen vaak de potentie, selectiviteit, of stabiliteit die nodig is om een medicijn te worden. Door de chemische structuur van deze leads te wijzigen, onderzoekers kunnen verbindingen ontwerpen met de juiste medicijnachtige eigenschappen. De ontworpen kandidaat-geneesmiddelen gaan vervolgens langs de ontwikkelingspijplijn naar de preklinische testfase. Van deze kandidaten slechts een klein deel komt in de klinische proeffase, en slechts één wordt uiteindelijk een goedgekeurd medicijn voor gebruik door patiënten. Het op de markt brengen van een nieuw medicijn kan tien jaar of langer duren en miljarden dollars kosten.
Knelpunten in het ontwerp van geneesmiddelen overwinnen door middel van computationele wetenschap
Recente ontwikkelingen in technologie en kennis hebben geresulteerd in een nieuw tijdperk van medicijnontdekking - een tijdperk dat de tijd en kosten van het ontwikkelingsproces van medicijnen aanzienlijk zou kunnen verminderen. Verbeteringen in ons begrip van de 3D-kristalstructuren van biologische moleculen en toename van rekenkracht maken het mogelijk om computationele methoden te gebruiken om interacties tussen geneesmiddelen te voorspellen.
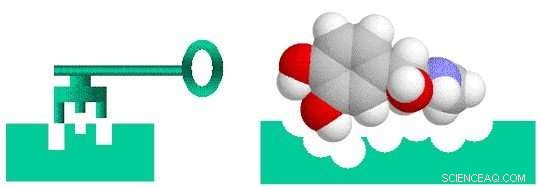
Drug discovery is een lock-and-key probleem waarbij het medicijn (sleutel) specifiek moet passen bij het biologische doelwit (slot). Krediet:Brookhaven National Laboratory
Vooral, een computersimulatietechniek genaamd moleculaire dynamica is veelbelovend gebleken in het nauwkeurig voorspellen van de sterkte waarmee medicijnmoleculen aan hun doelen binden (bindingsaffiniteit). Moleculaire dynamica simuleert hoe atomen en moleculen bewegen terwijl ze in hun omgeving interageren. In het geval van ontdekking van geneesmiddelen, de simulaties onthullen hoe medicijnmoleculen interageren met hun doeleiwit en de conformatie van het eiwit veranderen, of vorm, die zijn functie bepaalt.
Echter, deze voorspellingsmogelijkheden werken nog niet op een schaal die groot genoeg is of snel genoeg voor farmaceutische bedrijven om ze in hun ontwikkelingsproces te gebruiken.
"Om deze vooruitgang in voorspellende nauwkeurigheid te vertalen naar de industriële besluitvorming, moet in de orde van 10, 000 bindingsaffiniteiten worden zo snel mogelijk berekend, zonder verlies van nauwkeurigheid, " zei Jha. "Het produceren van tijdig inzicht vereist een rekenefficiëntie die is gebaseerd op de ontwikkeling van nieuwe algoritmen en schaalbare softwaresystemen, en de slimme toewijzing van supercomputerbronnen."
Jha en zijn medewerkers aan de Rutgers University, waar hij ook hoogleraar is aan de afdeling Electrical and Computer Engineering, en University College London ontwierpen een softwareraamwerk om de nauwkeurige en snelle berekening van bindingsaffiniteiten te ondersteunen en tegelijkertijd het gebruik van computerbronnen te optimaliseren. Dit kader, genaamd de High-Throughput Binding Affinity Calculator (HTBAC), bouwt voort op het RADICAL-Cybertools-project dat Jha leidt als hoofdonderzoeker van Rutgers' Research in Advanced Distributed Cyberinfrastructure and Applications Laboratory (RADICAL). Het doel van RADICAL-Cybertools is om een reeks softwarebouwstenen te bieden ter ondersteuning van de workflows van grootschalige wetenschappelijke toepassingen op krachtige computerplatforms, die rekenkracht bundelen om grote rekenproblemen op te lossen die anders onoplosbaar zouden zijn vanwege de benodigde tijd.
In de informatica, workflows verwijzen naar een reeks verwerkingsstappen die nodig zijn om een taak te voltooien of een probleem op te lossen. Vooral voor wetenschappelijke workflows, het is belangrijk dat de workflows flexibel zijn, zodat ze tijdens runtime dynamisch kunnen worden aangepast om de meest nauwkeurige resultaten te leveren en tegelijkertijd efficiënt gebruik te maken van de beschikbare rekentijd. Dergelijke adaptieve workflows zijn ideaal voor het ontdekken van geneesmiddelen, omdat alleen de geneesmiddelen met hoge bindingsaffiniteiten verder moeten worden geëvalueerd.
"De gewenste afweging tussen de vereiste nauwkeurigheid en de rekenkosten (tijd) verandert tijdens de ontdekking van het medicijn naarmate het proces van screening naar leadselectie en vervolgens leadoptimalisatie gaat, " zei Jha. "Een aanzienlijk aantal verbindingen moet goedkoop worden gescreend om slechte bindmiddelen te elimineren voordat er nauwkeurigere methoden nodig zijn om de beste bindmiddelen te onderscheiden. Om de snelste tijd-tot-oplossing te bieden, moet de voortgang van de simulaties worden bewaakt en beslissingen over voortgezette uitvoering worden gebaseerd op wetenschappelijke betekenis."
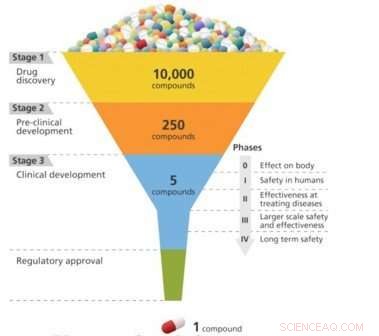
Een schematische weergave van het ontwikkelingsproces van geneesmiddelen, die geleidelijk de meest effectieve kandidaten uit een grote initiële pool aanscherpt. Krediet:Brookhaven National Laboratory
Met andere woorden, het zou geen zin hebben om de simulaties van een bepaalde geneesmiddel-eiwitinteractie voort te zetten als het geneesmiddel het eiwit zwak bindt in vergelijking met de andere kandidaten. Maar het zou logisch zijn om extra rekenkracht toe te wijzen als een medicijn een hoge bindingsaffiniteit vertoont.
Het ondersteunen van adaptieve workflows op grote schaal die kenmerkend zijn voor programma's voor het ontdekken van geneesmiddelen, vereist geavanceerde rekencapaciteiten. HTBAC biedt dergelijke ondersteuning via een flexibele middleware-softwarelaag die de adaptieve uitvoering van algoritmen mogelijk maakt. Momenteel, HTBAC ondersteunt twee algoritmen:verbeterde bemonstering van moleculaire dynamica met benadering van continuümoplosmiddel (ESMACS) en thermodynamische integratie met verbeterde bemonstering (TIES). ESMACS, een rekenkundig goedkopere maar minder rigoureuze methode dan TIES, berekent de bindingssterkte van een medicijn aan zijn doeleiwit op basis van moleculaire dynamische simulaties. Daarentegen, TIES vergelijkt de relatieve bindingsaffiniteiten van twee verschillende geneesmiddelen met hetzelfde eiwit.
"ESMACS biedt een snelle kwantitatieve benadering die gevoelig genoeg is om bindingsaffiniteiten te bepalen, zodat we slechte binders kunnen elimineren, terwijl TIES een nauwkeurigere methode biedt voor het onderzoeken van goede bindmiddelen omdat ze verfijnd en verbeterd zijn, " zei Jumana Dakka, een tweedejaars Ph.D. student aan Rutgers en lid van de groep RADICAL.
Om te bepalen welk algoritme moet worden uitgevoerd, HTBAC analyseert de bindingsaffiniteitsberekeningen tijdens runtime. This analysis informs decisions about the number of concurrent simulations to perform and whether stimulation steps should be added or removed for each drug candidate investigated.
Putting the framework to the test
Jha's team demonstrated how HTBAC could provide insight from drug candidate data on a short timescale by reproducing results from a collaborative study between University College London and the London-based pharmaceutical company GlaxoSmithKline to discover drug compounds that bind to the BRD4 protein. Known to play a key role in driving cancer and inflammatory diseases, the BRD4 protein is a major target of bromodomain-containing (BRD) inhibitors, a class of pharmaceutical drugs currently being evaluated in clinical trials. The researchers involved in this collaborative study are focusing on identifying promising new drugs to treat breast cancer while developing an understanding of why certain drugs fail in the presence of breast cancer gene mutations.
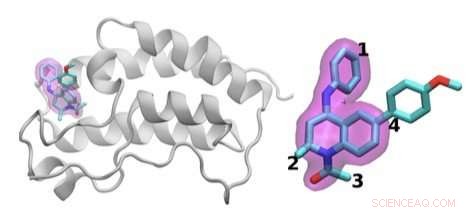
The scientists investigated the chemical structures of 16 drugs based on the same tetrahydroquinoline (THQ) scaffold. On the left is a cartoon of the BRD4 protein bound to one of these drugs; on the right is a molecular representation of a drug with the THQ scaffold highlighted in magenta. Regions that are chemically modified between the drugs investigated in this study are labeled 1 to 4. Typically, only a small change is made to the chemical structure of one drug to the next. This conservative approach makes it easier for researchers to understand why one drug is effective, whereas another is not. Credit:Brookhaven National Laboratory
Jha and his team concurrently screened a group of 16 closely related drug candidates from the study by running thousands of computational sequences on more than 32, 000 computing cores. They ran the computations on the Blue Waters supercomputer at the National Center for Computing Applications, University of Illinois at Urbana-Champaign.
In a real drug design scenario, many more compounds with a wider range of chemical properties would need to be investigated. The team members previously demonstrated that the workload management layer and runtime system underlying HTBAC could scale to handle 10, 000 concurrent tasks.
"HTBAC could support the concurrent screening of different compounds at unprecedented scales—both in the number of compounds and computational resources used, " said Jha. "We showed that HTBAC has the ability to solve a large number of drug candidates in essentially the same amount of time it would take to solve a smaller set, assuming the number of processors increases proportionally with the number of candidates."
This ability is made possible through HTBAC's adaptive functionality, which allows it to execute the optimal algorithm depending on the properties of the drugs being investigated, improving the accuracy of the results and minimizing compute time.
"The lead optimization stage usually considers on the order of 10, 000 small molecules, " said Jha. "While experiment automation reduces the amount of time needed to calculate the binding affinities, HTBAC has the potential to cut this time (and cost) by an order of magnitude or more."
With HTBAC, TIES requires approximately 25, 000 central processing unit (CPU) core hours for a single prediction. At least a 250 million core hours would be needed for a large-scale study to support a pharmaceutical drug screening campaign, with a typical turnaround time of about two weeks. HTBAC could facilitate running studies requiring sustained usage of millions of core hours per day.
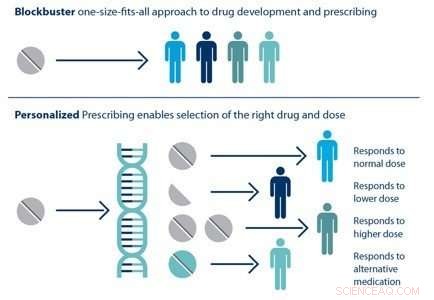
Individual patients respond differently to drugs. The ability to predict which treatment is best for a particular patient based on his or her genetic sequence is the goal of personalized medicine. Credit:Brookhaven National Laboratory
When the University of College London–GlaxoSmithKline study concludes, Jha and his team hope to be given the experimental data on the tens of thousands of drug candidates, without knowing which candidate ended up being the best one. Met deze informatie, they could perform a blind test to determine whether HTBAC provides an improvement in compute time (for a given accuracy) over the existing automated methods for drug discovery. Indien nodig, they could then refine their methodology.
Applying scalable computing to precision medicine
HTBAC not only has the potential to improve the speed and accuracy of drug discovery in the pharmaceutical industry but also to improve individual patient outcomes in clinical settings. Using target proteins based on a patient's genetic sequence, HTBAC could predict a patient's response to different drug treatments. This personalized assessment could replace the traditional one-size-fits-all approach to medicine. Bijvoorbeeld, such predictions could help determine which cancer patients would actually benefit from chemotherapy, avoiding unnecessary toxicity.
According to Jha, the computation time would have to be significantly reduced in order for physicians to clinically use HTBAC to treat their patients:"Our grand vision is to apply scalable computing techniques to personalized medicine. If we can use these techniques to optimize drugs and drug cocktails for each individual's unique genetic makeup on the order of a few days, we will be empowered to treat diseases much more effectively."
"Extreme-scale computing for precision medicine is an emerging area that CSI and Brookhaven at large have begun to tackle, " said CSI Director Kerstin Kleese van Dam. "This work is a great example of how technologies we originally developed to tackle DOE challenges can be applied to other domains of high national impact. We look forward to forming more strategic partnerships with other universities, pharmaceutical companies, and medical institutions in this important area that will transform the future of health care."
 Hoe de snelheid van een elektron te berekenen
Hoe de snelheid van een elektron te berekenen- Op weg naar de ontwikkeling van medicijnen voor aan veroudering gerelateerde ziekten
- Onderzoekers ontwikkelen nieuwe chemie om smartdrugs slimmer te maken
- Bewijsstuk A en andere echte misdaadshows kunnen misvattingen over forensische wetenschap aanwakkeren
- Nieuwe methode om olie uit water te verwijderen
- Nieuwe studies onderzoeken hoe kennis actie stimuleert bij de besluitvorming over klimaatverandering
- Hoe te vissen voor rivierkreeft en crawdads in Oregon
- NASA ziet een Picasso-achtig gezicht in tropische storm Cimarons krachtige stormen
- Is het maaien of kort grazen van weidegronden even heilzaam als voorgeschreven verbranding?
- Nieuwe studie toont aan dat vegetatie de toekomst van de watercyclus bepaalt
Hoofdlijnen
- Menselijke evolutie: tijdlijn, stadia, theorieën en bewijsmateriaal
- Race om Indonesische krokodil te redden die is getroffen door een bandenketting
- Wanneer stopt het leven op aarde?
- Maleisië omarmt het werk van Melanies aan de illegale handel in wilde dieren
- Onderzoekers ontdekken slecht begrepen bacteriële lijnen in de monden van dolfijnen
- Vliegtuigbrandstof uit suikerriet een realistisch vooruitzicht
- Waarom lijken oude stellen op elkaar?
- Nee,
- Structurele niveaus van organisatie van het menselijk lichaam
- Wetenschappers verbeteren computerweergave van dierenbont

- De strijd voor schone uitstoot gaat door

- Facebook start binnenkort met nominaties voor inhoudstoezichtspaneel

- Onderzoekers creëren AI-aangedreven chatbot om gezinnen met neurologische ontwikkelingsstoornissen te helpen

- Belgisch luchtruim gesloten na computerstoring


 Hoe een planeet in een computer te passen - het ontwikkelen van het energie-exascale aardesysteemmodel?
Hoe een planeet in een computer te passen - het ontwikkelen van het energie-exascale aardesysteemmodel?- Het samenvoegen van geheugen en berekening, programmeerbare chipsnelheden AI, verlaagt stroomverbruik
- In het moleculaire oog:vloeibare monsters in realtime onderzoeken
- Ontwerpen van hiërarchische nanoporeuze membranen voor zeer efficiënte adsorptie- en opslagtoepassingen
- Hoe gouden nanodeeltjes de opslag van zonne-energie kunnen verbeteren
- Hoe kunnen we het smelten van gletsjers stoppen?
- Hoe retailers meer geld kunnen verdienen in online veilingen
- Toenemende opsluiting van First Nations-vrouwen is verweven met de ervaring van geweld en trauma
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
