Wetenschap
Machine learning en AI helpen bij het voorspellen van de moleculaire selectiviteit van chemische reacties
Er zijn nu weinig problemen die AI en machinaal leren niet kunnen helpen oplossen. Onderzoekers van de Yokohama National University gebruiken dit moderne voordeel om op te lossen wat conventionele methoden niet kunnen.
Er zijn veel regels die je moet onthouden als het gaat om de interactie van koolstofhoudende (of organische) moleculen:de positionering van groepen op het molecuul die interageren met zijn omgeving, de grootte, vorm en positie van het molecuul, en het molecuul waarmee het is interactie. De uitkomst van een bepaalde reactie kan heel verschillend zijn, afhankelijk van deze en nog veel meer factoren, en het voorspellen van deze uitkomsten is op chemisch gebied een behoorlijke uitdaging gebleken. Het beheersen van de uitkomst is een zeer noodzakelijk onderdeel van de chemische synthese, maar voorspellingen zijn niet altijd voldoende.
Gelukkig kunnen machinaal leren en kunstmatige intelligentie (AI) opnieuw de vooruitgang helpen bevorderen door de snelheid of selectiviteit van een bepaalde reactie te voorspellen. Daarom kan deze technologie nuttig zijn bij het voorspellen welk product kan worden verwacht.
De onderzoekers hebben hun bevindingen gepubliceerd in het Journal of Chemical Information and Modeling .
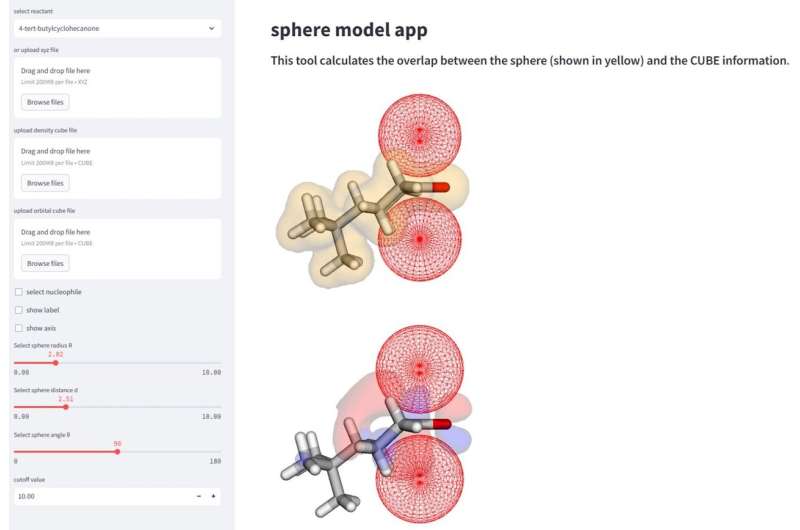
In de organische chemie is elk detail van belang. Twee gemeenschappelijke gebieden die van invloed kunnen zijn op de manier waarop een molecuul met andere moleculen communiceert, zijn sterische en orbitalen. Sterische stoffen hebben betrekking op de rangschikking van moleculen en sterische effecten kunnen de vorm en reactiviteit van het molecuul bepalen. Dit kan te wijten zijn aan de grootte of lading van het molecuul of het individuele atoom. Orbitalen zijn een manier om de meest waarschijnlijke plaatsing van de elektronen te verklaren, die op hun beurt kunnen interageren met andere moleculen of atomen om reacties te veroorzaken.
Deze factoren kunnen drastisch veranderen waar een nucleofiel, of een elektronendonerende reactant, zich kan hechten aan het ontvangende molecuul. Dit staat bekend als "selectiviteit", en afhankelijk van waar het molecuul zich hecht, kunnen de resultaten verschillende producten of opbrengsten van het gewenste product vormen. Onderzoekers gebruiken AI en machinaal leren, evenals de huidige kennis van chemische reacties, om deze aspecten van moleculaire selectiviteit beter te verklaren.
"Om te bepalen welke informatie kan worden gebruikt als essentiële chemische informatie die aan AI moet worden gegeven, is het noodzakelijk om chemische kennis te combineren met kennis van AI en machinaal leren", zegt de overeenkomstige auteur Hiroaki Gotoh, universitair hoofddocent aan de faculteit Ingenieurswetenschappen, Yokohama. Nationale Universiteit.
Eerst moest de computer wat informatie krijgen waaruit hij kon leren. Informatie uit de literatuur op het gebied van computationele chemie en informatie uit eerdere onderzoeken werd gebruikt om het leerproces van de AI te starten. Na enige handmatige gegevensinvoer voor de specifieke moleculen die werden gebruikt en het instellen van optimale parameters, werden gegevensanalyses uitgevoerd op basis van de voorspelde resultaten van de testgegevensset. Dankzij deze analyses kan de AI toekomstige selectiviteiten leren en voorspellen op basis van reeds bekende informatie.
"Deze methode maakt een uitgebreidere analyse en interpretatie van reactiemechanismen mogelijk via berekening van de parameters van de bolvormige ruimtes die naderende nucleofielen nabootsen", zegt Daimon Sakaguchi, eerste auteur van de studie aan de afdeling scheikunde en levenswetenschappen van de Yokohama National University.
De studie verklaarde met succes de reactieselectiviteit van acht nucleofielen, op basis van welke "zijde" van het molecuul de meest wenselijke hoeveelheid product zou opleveren. De selectiviteit verandert op basis van de sterics van het molecuul, naast de orbitale factoren. Onderzoekers ontdekten dat voor sommige moleculen de orbitale factor belangrijker is bij het bepalen van de gezichtsselectiviteit, en dat andere meer afhankelijk zijn van de sterische eigenschappen van het molecuul wanneer het interageert met zijn nucleofiel.
De combinatie van voorspellende technologie en machinaal leren met gevestigde kennis van de chemie kan betere resultaten opleveren uit de chemische reactie en scheikundigen helpen natuurlijke producten en farmaceutische chemicaliën op een meer gestroomlijnde manier te synthetiseren.
Door dit proces te stroomlijnen met het gebruik van machine learning en kunstmatige intelligentie, kunnen er meer experimenten plaatsvinden. Idealiter hopen de onderzoekers samen te werken met experimentele scheikundigen om reacties te ontwerpen die zullen doorgaan met de ontwikkeling van meer voorspellende technologie voor chemische reacties.