Wetenschap
Supercomputersimulaties onthullen de details van coronavirusfusie
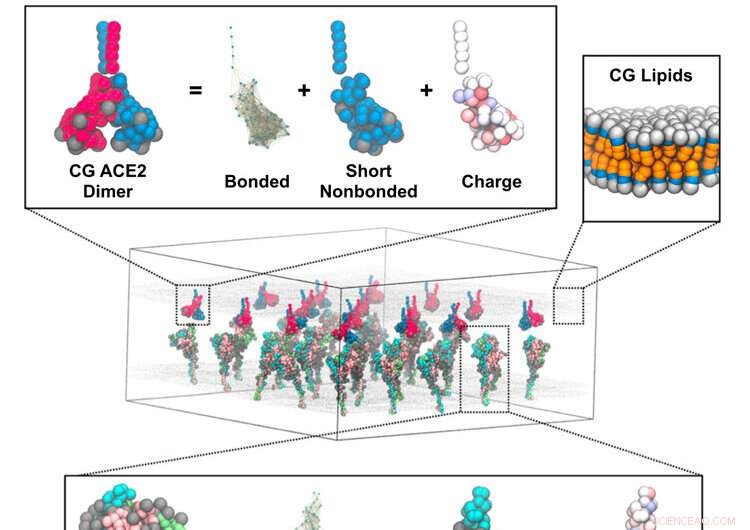
Het mechanisme waarmee het coronavirus fuseert met gastheercellen is gesuggereerd door simulaties door onderzoekers van de Universiteit van Chicago met behulp van de Frontera-supercomputer van TACC. Representatieve afbeelding van een cursus-grained (CG) simulatie van spike trimeren in membraan interactie met een aangrenzend membraan met ACE2 dimeren. De inzetstukken tonen de CG-modelcomponenten voor de spike-trimeer (onder), ACE2-dimeer (linksboven) en lipidemembraan (rechtsboven). Credit:Pak, A.J., Yu, A., Ke, Z. et al.
Het mysterie van hoe het SARS-CoV-2-virus menselijke longcellen precies infecteert, blijft grotendeels verborgen voor experimentele wetenschappers. Nu zijn echter de duivelse details van het mechanisme waarmee het coronavirus samensmelt met gastheercellen, gesuggereerd door simulaties door onderzoekers van de Universiteit van Chicago met behulp van de Frontera-supercomputer in het Texas Advanced Computing Center (TACC).
De computermodellen tonen het coöperatieve gedrag van gastheercelreceptoreiwitten dat leidt tot hun eigen infectie. Het werk kan worden toegepast om de toegenomen virulentie van coronavirusvarianten zoals delta, omicron en meer te helpen begrijpen.
"We ontdekten dat het spike-eiwit op een zeer coöperatieve manier interageert met twee ACE2-receptoren", zegt Gregory Voth, een vooraanstaande professor in de chemie aan de Universiteit van Chicago. "Dit is een fundamenteel biofysisch inzicht."
Voth is hoofdauteur van de studie die de interacties tussen het coronavirus en de receptorcellen heeft gemodelleerd met computersimulaties, gepubliceerd in het tijdschrift Nature Communications in februari 2022.
Als een voetbal met spikes sieren de spike-eiwitten het oppervlak van het coronavirus. De spikes zoeken en fuseren met de angiotensine-converting enzyme 2 (ACE2) eiwitreceptoren in menselijke longcellen. Het spike-eiwit bestaat uit twee hoofdonderdelen. Het S1-domein bevat het receptorbindende domein dat ACE2-eiwitten herkent. En het S2-domein bevat de fusiemachinerie, die als een schil wordt beschermd en bedekt door het S1-domein.
De simulaties laten zien hoe het ene ACE2-receptoreiwit de coronaviruspiek vasthoudt en verzwakt, terwijl het andere het uit elkaar begint te trekken. Het S1-domein valt dan uit elkaar en stelt de fusiemachinerie bloot. Deze "een-twee"-stoot bereidt het virus voor op fusie en toegang tot menselijke longgastcellen.
"Het lijkt erop dat varianten zoals delta en omicron dat gedrag nog meer kunnen accentueren - het is een belangrijke stap. Uiteindelijk moeten toekomstige antilichamen en mogelijk moleculaire geneesmiddelen dit proces kunnen verstoren," zei Voth.
Voth en collega's ontwikkelden wat zij "bottom-up grofkorrelige modellen" noemen, die cryo-elektronentomografiegegevens van het laboratorium van studie co-auteur John Briggs van het Max Planck Institute of Biochemistry gebruikten. Ze combineerden het met atomistische simulaties van moleculaire dynamica. De gegenereerde gegevens werden ingevoerd in een theoretisch kader dat de grofkorrelige modellen ontwikkelde.
"De grofkorrelige modellen zijn tot 1000 keer sneller dan directe atomistische moleculaire dynamica-simulaties, maar ze behouden de essentiële fysieke kenmerken," zei Voth. Deze methode levert een enorme besparing op in tijd en geld op de berekeningen.
Het wetenschappelijke team ontving supercomputerbronnen en -diensten van het COVID-19 HPC Consortium, een publiek-private inspanning ter ondersteuning van COVID-19-onderzoek. Via het consortium gebruikten ze het door de National Science Foundation gefinancierde Frontera-systeem bij TACC; het Witherspoon-computercluster bij IBM Research; en middelen van de Oak Ridge Leadership Computing Facility in het Oak Ridge National Laboratory.
"We hebben moleculaire dynamica-gegevens van alle atomen op Frontera berekend en analysetools gebruikt die beschikbaar zijn bij TACC - beide waren zeer waardevol," zei Voth.
Het team van Voth diende hun paper in voordat de delta- en ommicronvarianten bekend waren, en voorspelde daarom de mutaties niet. Maar ze zijn wel teruggegaan en hebben de modellen herzien om de varianten te onderzoeken.
"Delta heeft zoiets als een opening in de piek die gemakkelijker optreedt dan bij eerdere coronavirusmutaties," zei Voth. "Het voelde opwindend vanuit wetenschappelijk oogpunt om gedrag te zien dat nog niet eerder was gezien."
Voth verwees naar laboratoriumgegevens voor cryo-elektronenmicroscopie die de structuur van een oplosbaar spike-eiwit met twee ACE2-receptoren eraan gebonden laten zien. Maar hij onderscheidde dit uitgekristalliseerde voorbeeld van wat hij onderzocht met behulp van simulaties in de meer realistische omgeving van veel eiwitten die op membraanplaten met elkaar in wisselwerking staan.
"Supercomputers kunnen, mits goed gebruikt en gebaseerd op goede fysica, een geheel nieuwe manier bieden om naar deze processen te kijken. Door computersimulatie kan men dingen bestuderen die momenteel niet met experimenten kunnen worden gedaan. Simulatie en experimenten werken heel goed samen, hand in hand," zei Voth. + Verder verkennen
Eerste complete coronavirusmodel toont samenwerking
 Een minerale spons gebruiken om uranium op te vangen
Een minerale spons gebruiken om uranium op te vangen- Techniek om fosfor uit urine terug te winnen
- Nieuw proces maakt biologisch afbreekbare kunststoffen echt composteerbaar
- Wat zit er in een permanente marker?
- We huwen wanorde met orde:de effecten van geometrische wanorde op vloeistoffen en vaste stoffen in mesoporeuze materialen
- Vulkaan Bali spuwt rook en as, angst voor uitbarsting opwekken
- Hoe de La Brea-teerputten werken
- Reductie van methaanemissies uit meren mogelijk met nieuwe aanpak
- Beter veilig dan sorry - economische optimalisatie riskeert kantelen van elementen van het aardsysteem
- Crowdsourced stamboom levert nieuwe inzichten over de mensheid op
Hoofdlijnen
- Processen die ATP als energiebron gebruiken
- Invasieve habitat voor stinkwantsen kan enorm uitbreiden door klimaatverandering
- Nieuwe biobronnen voor plantaardige peptidehormonen met behulp van technologie voor het bewerken van genen
- Wanneer treedt melkzuurfermentatie op?
- Naar huis vliegen? IJstijd heeft de vogeltrek mogelijk afgeremd
- Nieuwe kaarten laten zien dat de krimpende wildernis op eigen risico wordt genegeerd
- Oplossen van hoe een complexe ziekte eiken bedreigt
- Onderzoekers vinden eerste wilde alligator brekende schildpad in Illinois sinds 1984
- Hoe nemen we beslissingen?




 Moleculaire steigers helpen constructie op nanoschaal
Moleculaire steigers helpen constructie op nanoschaal- Ras, etniciteit geen factor in recent wapendragend gedrag op Amerikaanse scholen
- Boeing verwacht niet dat het testprobleem de 777X . zal vertragen
- Wat zijn de toepassingen van een destillatiekolf?
- Onderzoekers reconstrueren het genoom van de gemeenschappelijke voorouder van alle zoogdieren
- Koolstofnanobuisjes openen nieuwe horizonten voor neurowetenschap:controle van de uitgroei van neurale cellen
- Beleidsvorming op het gebied van vervoer in Chinese steden
- Hoe de pandemie van het coronavirus de perfecte storm van Florida werd
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
