Wetenschap
Moleculaire simulaties laten zien hoe medicijnen belangrijke receptoren blokkeren

Krediet:CC0 Publiek Domein
Veel geneesmiddelen werken door zich te richten op wat bekend staat als 'G-proteïne-gekoppelde receptoren'. In een nieuwe studie, wetenschappers van de Universiteit van Uppsala beschrijven hoe ze hebben kunnen voorspellen hoe speciale moleculen die kunnen worden gebruikt in nieuwe immunotherapie tegen kanker zich binden aan deze receptoren. De rekenmethoden van de onderzoekers, gepresenteerd in het tijdschrift Angewandte Chemie zijn een essentiële bijdrage aan het toekomstige, op structuren gebaseerde medicijnontwerp.
G-eiwit-gekoppelde receptoren (GPCR's) behoren tot de eiwitdoelgroepen die van het grootste belang zijn voor de ontwikkeling van geneesmiddelen. Deze receptoren reageren op bijvoorbeeld, licht, smaken, geuren, adrenaline, histamine, dopamine en een lange lijst van andere moleculen door verdere biochemische signalen in cellen door te geven. De onderzoekers die het onderzoek naar GPCR's uitvoerden, werden in 2012 beloond met de Nobelprijs voor de Scheikunde.
Vandaag, ongeveer 30 procent van alle geneesmiddelen op de markt hebben GPCR's als hun doeleiwitten. Sommige medicijnmoleculen, zoals morfine, activeren de receptoren (agonisten), terwijl andere, zoals bètablokkers, inactiveren (antagonisten).
Een belangrijke GPCR is de adenosine A2A-receptor. De antagonisten ervan kunnen worden gebruikt in nieuwe immunotherapie tegen kanker. Samen met het biofarmaceutische bedrijf Sosei-Heptares, de onderzoekers Willem Jespers, Johan Åqvist en Hugo Gutierrez-de-Terán van de Universiteit van Uppsala zijn erin geslaagd te laten zien hoe een reeks A2A-antagonisten zich aan de receptor binden en deze inactiveren.
Met moleculaire dynamische simulaties en berekening van bindingsenergieën, het werd mogelijk te voorspellen hoe moleculen van het farmaceutisch bedrijf zich aan de receptoren zouden binden en hoe sterk ze dat doen. Daarna, nieuwe antagonisten werden ontworpen, en gesynthetiseerd door chemici van de universiteit van Santiago de Compostela, Spanje. Driedimensionale structuren van de complexen die zich vormen tussen deze moleculen en de receptor werden vervolgens experimenteel bepaald met röntgenkristallografie. Computerberekeningen bleken in staat om zowel de structuur als de bindingssterkte in de complexen met grote precisie te voorspellen.
"Dit is een stevige stap voorwaarts, en we hebben met grote precisie kunnen voorspellen hoe deze familie van moleculen de A2A-receptor bindt. Onze berekeningsmethoden kennen nu een grote doorbraak in het op structuren gebaseerde medicijnontwerp, " zegt Hugo Gutierrez-de-Terán, die het project van de Uppsala-groep leidde.

Hoofdlijnen
- Hoe is DNA geordend om in een cel te passen?
- Verschil tussen transcriptie en DNA-replicatie
- Kenmerken van nucleïnezuren
- Het combineren van cassavemeel in het brooddeeg kan in de toekomst de toegang tot voedsel verzekeren
- Het belang van het bestuderen van de DNA-genetica van het menselijk geslacht
- Wat is het geheim van geluk?
- Hoe snel is een knipoog?
- De celstructuur van een ui
- Helpers bij het nest kunnen moedervogels toestaan kleinere eieren te leggen
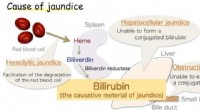
- Onderzoekers maken doorbraak in antioxidant-enzym gekoppeld aan geelzucht
- Ontwikkeling van vereenvoudigde nieuwe massaspectrometrische techniek met behulp van laser en grafeen

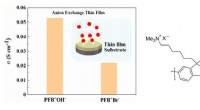
- De eerste geleidbaarheid van hydroxide in dunne films van aniongeleidende polymeren
- Ontdekking kan microverontreinigingen uit het milieu verwijderen

- Vastestofelektrolyt met lithium-iongeleidbaarheid vergelijkbaar met vloeibare elektrolyten


 Er waren enkele onwaarschijnlijke maar zeer gelukkige overlevenden van de Notre Dame Fire
Er waren enkele onwaarschijnlijke maar zeer gelukkige overlevenden van de Notre Dame Fire- Flushed Goldfish neemt de grote meren over - Ja, echt waar!
- Nieuwe bio-geïnspireerde hydrogels kunnen werken als superlijm in zeer ionische omgevingen zoals zeewater
- Microplastics beïnvloeden de wereldwijde nutriëntencyclus en het zuurstofgehalte in de oceaan
- KSU-team draagt bij aan DUNE, 's werelds grootste neutrino-experiment
- Effecten van ijstijden uit het verleden wijdverbreid dan eerder werd gedacht
- Things Made From Recycled Plastic
- Hoe te bepalen of de binding tussen twee atomen polair is?
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
