Wetenschap
Moleculaire dynamische simulatie werpt nieuw licht op de vorming van methaanhydraat

Methaanhydraat gewonnen uit de oceaanbodem voor de kust van Oregon, VS. Krediet:Wikimedia Commons
In een artikel dat deze week is gepubliceerd in PNAS , onderzoekers van het Van 't Hoff Instituut voor Moleculaire Wetenschappen van de Universiteit van Amsterdam en het Amsterdam Centre for Multiscale Modeling geven atomistisch inzicht in de vorming van methaanhydraten. Op basis van moleculaire dynamica-simulaties leggen ze uit hoe selectie tussen concurrerende methaanhydraatpolymorfen plaatsvindt, en hoe dit kan worden gegeneraliseerd naar andere hydraten en moleculaire kristalvorming.
Methaanhydraat zijn ijsachtige vaste stoffen die overvloedig aanwezig zijn, onder andere op de oceaanbodem. Geschat wordt dat de hoeveelheid energie die is opgeslagen in methaanhydraten twee keer zo groot is als de hoeveelheid energie die is opgeslagen in conventionele bronnen van fossiele brandstoffen. Tegelijkertijd, de vorming van hydraten is een bron van zorg voor de aardolie-industrie omdat ze oliepijpleidingen kunnen verstoppen, doorstromingsproblemen veroorzaken. Methaanhydraten zijn ook aanwezig in de permafrost in arctische gebieden. Het ontdooien van de permafrost als gevolg van de stijgende wereldtemperaturen kan leiden tot het vrijkomen van grote hoeveelheden methaan, wat een krachtig broeikasgas is.
Ingekapselde methaanmoleculen
In een methaanhydraat, op moleculair niveau zit methaan opgesloten in een waterstofgebonden waternetwerk. Terwijl methaangas hydrofoob is onder omgevingsomstandigheden, bij lage temperaturen en hoge drukken kan een mengsel van water en methaangas spontaan kiemen tot hydraten.
Door de jaren heen, de interesse in het begrijpen van het vormingsmechanisme van hydraten is enorm toegenomen. Met name hun vorming onder natuurlijke omstandigheden is slecht begrepen. Het proces van homogene kiemvorming begrijpen, en hoe dit leidt tot verschillende methaanhydraatpolymorfen, kan leiden tot een betere controle van de kristallisatie, evenals inzicht in polymorfe selectie in het algemeen.
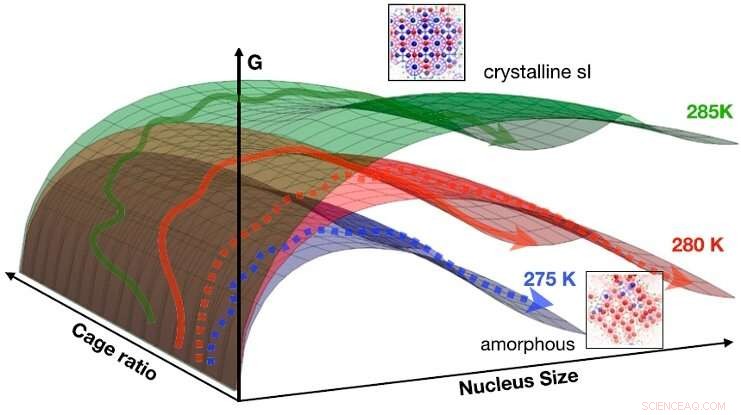
De bevindingen kunnen worden samengevat in een geïdealiseerd CNT-oppervlak met vrije energie als functie van de grootte en de kooiverhouding voor 275 K (blauw), 280 K (rood), en 285 K (groen). De pijlen geven schematisch paden aan die lopen van vloeistof naar vaste stof (gestippelde pijlen:naar amorfe fase; vaste pijlen:naar kristallijne fase). Bij lage temperaturen (bijv. 275K), de vrije energiebarrière om de amorfe vaste stof te kiemen is het laagst, de trend wordt omgekeerd bij hogere temperaturen (bijv. 285K), waar bemonsterde paden meestal in de kristallijne fase terechtkomen. Bij 280 K zijn beide mechanismen toegankelijk. Krediet:HIMS/PNAS
Nieuwe simulatiebenadering
Omdat experimenteel onderzoek naar de vorming van de verschillende methaanhydraatpolymorfen een beperkte resolutie heeft, de Amsterdamse onderzoekers onder leiding van professor Peter Bolhuis gebruikten moleculaire dynamica-simulaties om dat inzicht te verschaffen.
Het toepassen van een directe moleculaire dynamica-simulatie is niet erg effectief, omdat bij matige onderkoeling kiemvorming een zeer zeldzame gebeurtenis is, door de aanwezigheid van een zeer hoge energiebarrière. Een dergelijke simulatie zou rekentijden vergen die verder gaan dan de leeftijd van het heelal. Echter, omdat de kiemvorming op zichzelf, hoewel zeldzaam, gebeurt zeer snel (op een microseconde tijdschaal), de onderzoekers zouden een grote verzameling moleculaire dynamica-trajecten kunnen creëren die deze snelle gebeurtenissen vertonen. Daaropvolgende gedetailleerde analyse van deze trajecten toonde aan hoe selectie tussen concurrerende amorfe en kristallijne polymorfe vormingsmechanismen plaatsvindt. Hun PNAS-papier werpt niet alleen licht op de vorming van methaanhydraten, maar ook op andere clathraatverbindingen en moleculaire kristalvorming in het algemeen.
 DNA is slechts één van de miljoenen mogelijke genetische moleculen
DNA is slechts één van de miljoenen mogelijke genetische moleculen- De verschillen tussen zout en suiker
- Kalkoenborst met een lager natriumgehalte wint sensorische test dan volzoutoptie
- Nanokristallen worden beter als ze verdubbelen met MOF's
- Chemici synthetiseren elektroden voor accu's uit koffiedik
- NASA vindt nog een klein gebied met sterke stormen in vervagende Flossie
- De epische zoektocht naar het oudste ijs op Antarctica
- Financiering voor klimaatadaptatie is ineffectief en moet transparanter
- Het is meer dan alleen klimaatverandering
- Burgers en wetenschappers publiceren 28-jarig record van waterkwaliteit in Buzzards Bay
Hoofdlijnen
- De cyanideverdediging:hoe één bacterie roofdieren remt met gif
- Hoe de botten in het menselijk skelet te bestuderen
- Waarom zijn cellen belangrijk voor levende organismen?
- Hoe vergif Sumac te behandelen?
- Waar komt collageen vandaan?
- Hoe goede bacteriën je genen beheersen
- Massa-extincties verwijderen soorten, maar geen ecologische variëteit
- Wat gebruiken chloroplasten om glucose te maken?
- Plant: definitie, evolutie, taxonomie
- structurele kleuren, zonder de glans

- Krakende ontdekking:Japanse wetenschapper gebruikt eiwit voor schone energie

- MXene-materialen helpen fotodetectoren het licht te zien

- Plastic transparanter maken en tegelijkertijd elektrische geleidbaarheid toevoegen

- Wetenschappelijke literatuur over oxidatieve reacties geanalyseerd


 Nanocrack-coating zorgt ervoor dat membranen bij hoge temperaturen kunnen werken, omgevingen met een lage luchtvochtigheid
Nanocrack-coating zorgt ervoor dat membranen bij hoge temperaturen kunnen werken, omgevingen met een lage luchtvochtigheid- Testen van de antibacteriële eigenschappen van hydrofobe oppervlakken op het ISS
- Layer-cake 2-D supergeleiding:het ontwikkelen van schone 2-D supergeleiding in een bulk van der Waals superrooster
- Bij de uitfasering van kolen, Duitsland zegt miljarden toe aan mijnbouwregio's
- In het oude Mesopotamië, seks tussen de goden schudde hemel en aarde
- Insecten die leven Underground
- Negatieve getallen converteren naar binair
- Waarom Elon Musk het bij het verkeerde eind heeft over nanotechnologie
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
