Wetenschap
Nieuwe tool decodeert complexe genomische gegevens van één cel
Het ontsluiten van biologische informatie uit complexe eencellige genomische gegevens is zojuist eenvoudiger en preciezer geworden, dankzij de innovatieve scLENS-tool ontwikkeld door de Biomedical Mathematics Group binnen het IBS Center for Mathematical and Computational Sciences onder leiding van hoofdonderzoeker Kim Jae Kyoung, die ook een professor aan KAIST. Dit betekent een aanzienlijke sprong voorwaarts op het gebied van single-cell transcriptomics.
Het onderzoek is gepubliceerd in het tijdschrift Nature Communications .
Eencellige genomische analyse is een geavanceerde techniek die genexpressie op individueel celniveau meet, waardoor cellulaire veranderingen en interacties aan het licht komen die niet waarneembaar zijn met traditionele genomische analysemethoden. Wanneer deze analyse wordt toegepast op kankerweefsels, kan deze de samenstelling van diverse celtypen binnen een tumor in kaart brengen, waardoor inzicht wordt verkregen in de voortgang van kanker en de belangrijkste genen worden geïdentificeerd die betrokken zijn bij elke fase van de progressie.
Ondanks het enorme potentieel van eencellige genomische analyse, is het omgaan met de enorme hoeveelheid gegevens die het genereert altijd een uitdaging geweest. De hoeveelheid gegevens omvat de expressie van tienduizenden genen in honderden tot duizenden individuele cellen. Dit resulteert niet alleen in grote datasets, maar introduceert ook ruisgerelateerde vervormingen, die gedeeltelijk ontstaan als gevolg van de huidige meetbeperkingen.
De corresponderende auteur Kim Jae Kyoung benadrukte:"Er is de afgelopen tien jaar een opmerkelijke vooruitgang geboekt in experimentele technologieën voor het analyseren van transcriptomen van één cel. Vanwege beperkingen in de methoden voor gegevensanalyse is er echter moeite geweest om waardevolle gegevens die zijn verkregen via uitgebreide kosten en tijd."
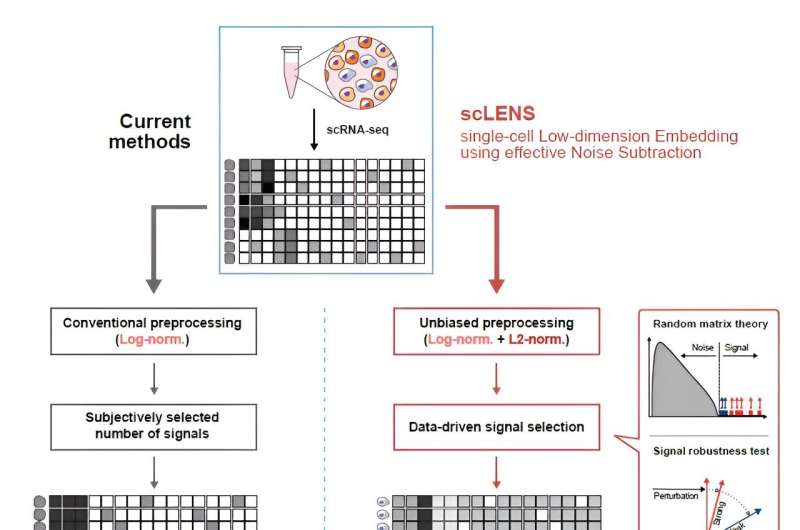
Onderzoekers hebben in de loop der jaren talloze analysemethoden ontwikkeld om biologische signalen uit deze ruis te onderscheiden. De nauwkeurigheid van deze methoden was echter niet bevredigend. Een cruciaal probleem is dat het bepalen van signaal- en ruisdrempels vaak afhangt van subjectieve beslissingen van de gebruikers.
De nieuw ontwikkelde scLENS-tool maakt gebruik van Random Matrix Theory en signaalrobuustheidstests om automatisch signalen van ruis te onderscheiden zonder afhankelijk te zijn van subjectieve gebruikersinvoer.
Eerste auteur Kim Hyun verklaarde:"Voorheen moesten gebruikers willekeurig de drempelwaarde voor signaal en ruis bepalen, wat de reproduceerbaarheid van de analyseresultaten in gevaar bracht en subjectiviteit introduceerde. scLENS elimineert dit probleem door automatisch signalen te detecteren met alleen de inherente structuur van de gegevens."
Tijdens de ontwikkeling van scLENS hebben onderzoekers de fundamentele redenen voor onnauwkeurigheden in bestaande analysemethoden geïdentificeerd. Ze ontdekten dat veelgebruikte methoden voor gegevensvoorverwerking zowel biologische signalen als ruis vervormen. De nieuwe voorverwerkingsaanpak die scLENS biedt, is vrij van dergelijke vervormingen.
Door problemen op te lossen die verband houden met de ruisdrempel bepaald door subjectieve gebruikerskeuze en signaalvervorming bij conventionele gegevensvoorverwerking, presteert scLENS aanzienlijk beter dan bestaande methoden op het gebied van nauwkeurigheid. Bovendien automatiseert scLENS het moeizame proces van signaaldimensieselectie, waardoor onderzoekers biologische signalen gemakkelijk en automatisch kunnen extraheren.
Ci Kim voegde eraan toe:"scLENS lost grote problemen op bij de analyse van transcriptoomgegevens van eencellige cellen, waardoor de nauwkeurigheid en efficiëntie tijdens het hele analyseproces aanzienlijk wordt verbeterd. Dit is een goed voorbeeld van hoe fundamentele wiskundige theorieën innovatie in biowetenschappelijk onderzoek kunnen stimuleren, waardoor onderzoekers meer kunnen doen beantwoord snel en nauwkeurig biologische vragen en ontdek geheimen van het leven die voorheen verborgen waren."