Wetenschap
Genetische analyses onthullen nieuwe virussen aan de horizon
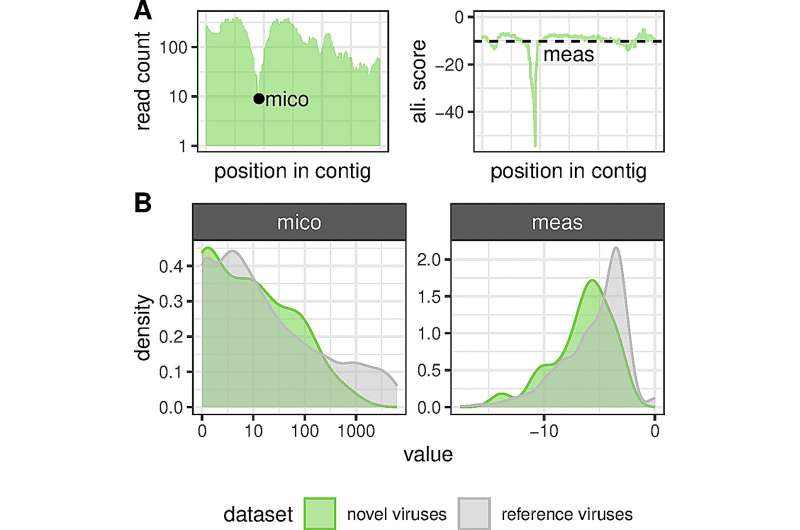
Plots verschijnen ze, en net als het SARS-CoV-2-coronavirus kunnen ze grote epidemieën veroorzaken:virussen die niemand op hun radar had. Ze zijn niet echt nieuw, maar ze zijn genetisch veranderd. Met name de uitwisseling van genetisch materiaal tussen verschillende virussoorten kan leiden tot de plotselinge opkomst van bedreigende ziekteverwekkers met aanzienlijk gewijzigde kenmerken.
Dit blijkt uit huidige genetische analyses die zijn uitgevoerd door een internationaal team van onderzoekers. Virologen van het Duitse Kankeronderzoekscentrum (DKFZ) hadden de leiding over het grootschalige onderzoek, gepubliceerd in het tijdschrift PLOS Pathogens .
“Met behulp van een nieuwe computerondersteunde analysemethode hebben we 40 voorheen onbekende nidovirussen ontdekt bij verschillende gewervelde dieren, van vissen tot knaagdieren, waaronder 13 coronavirussen”, meldt DKFZ-groepsleider Stefan Seitz. Met behulp van krachtige computers heeft het onderzoeksteam, waartoe ook de werkgroep van Chris Lauber van het Helmholtz Center for Infection Research in Hannover behoort, bijna 300.000 datasets doorzocht. Dat we nu zulke grote hoeveelheden data in één keer kunnen analyseren, opent volgens viroloog Seitz compleet nieuwe perspectieven.
Virusonderzoek staat nog in de relatieve kinderschoenen. Er is slechts een fractie bekend van alle virussen die in de natuur voorkomen, vooral de virussen die ziekten veroorzaken bij mensen, huisdieren en gewassen. De nieuwe methode belooft daarom een kwantumsprong in de kennis met betrekking tot het natuurlijke virusreservoir. Stefan Seitz en zijn collega's stuurden via hun krachtige computers genetische gegevens van gewervelde dieren die in wetenschappelijke databases waren opgeslagen, met nieuwe vragen. Ze zochten naar met virus geïnfecteerde dieren om op grote schaal viraal genetisch materiaal te verkrijgen en te bestuderen. De nadruk lag vooral op zogenaamde nidovirussen, waartoe ook de coronavirusfamilie behoort.
Nidovirussen, waarvan het genetische materiaal uit RNA (ribonucleïnezuur) bestaat, zijn wijdverspreid bij gewervelde dieren. Deze soortrijke groep virussen heeft enkele gemeenschappelijke kenmerken die hen onderscheiden van alle andere RNA-virussen en hun relatie documenteren. Voor het overige verschillen nidovirussen echter sterk van elkaar, d.w.z. in termen van de grootte van hun genoom.
Eén ontdekking is vooral interessant met betrekking tot de opkomst van nieuwe virussen:bij gastheerdieren die tegelijkertijd met verschillende virussen zijn geïnfecteerd, kan tijdens de virusreplicatie een recombinatie van virale genen optreden.
"Blijkbaar wisselen de nidovirussen die we bij vissen hebben ontdekt regelmatig genetisch materiaal uit tussen verschillende virussoorten, zelfs over familiegrenzen heen", zegt Seitz. En wanneer verre familieleden ‘kruisen’, kan dit leiden tot de opkomst van virussen met geheel nieuwe eigenschappen. Volgens Seitz kunnen dergelijke evolutionaire sprongen de agressiviteit en gevaarlijkheid van de virussen beïnvloeden, maar ook hun gehechtheid aan bepaalde gastdieren.
"Een genetische uitwisseling, zoals we hebben gevonden bij visvirussen, zal waarschijnlijk ook plaatsvinden bij zoogdiervirussen", legt Seitz uit. Vleermuizen, die net als spitsmuizen vaak besmet zijn met een groot aantal verschillende virussen, worden beschouwd als een echte smeltkroes. Het SARS-CoV-2 coronavirus heeft zich waarschijnlijk ook ontwikkeld in vleermuizen en is van daaruit overgesprongen naar de mens.
Na genuitwisseling tussen nidovirussen verandert vaak het spike-eiwit waarmee de virussen zich aan hun gastheercellen hechten. Chris Lauber, eerste auteur van het onderzoek, kon dit aantonen door middel van stamboomanalyses. Het aanpassen van dit ankermolecuul kan de eigenschappen van de virussen aanzienlijk in hun voordeel veranderen, door hun besmettelijkheid te vergroten of ze in staat te stellen van gastheer te wisselen.
Een verandering van gastheer, vooral van dier naar mens, kan de verspreiding van het virus sterk vergemakkelijken, zoals de coronapandemie nadrukkelijk heeft aangetoond. Virale ‘game-changers’ kunnen op elk moment plotseling opduiken en een enorme bedreiging worden, en – als het erop aankomt – een pandemie veroorzaken. Het uitgangspunt kan een enkel dubbel besmet gastdier zijn.
Het nieuwe krachtige computerproces zou de verspreiding van nieuwe virussen kunnen helpen voorkomen. Het maakt een systematische zoektocht naar virusvarianten mogelijk die potentieel gevaarlijk zijn voor de mens, legt Seitz uit.
De DKFZ-onderzoeker ziet nog een belangrijke mogelijke toepassing met betrekking tot zijn bijzondere onderzoeksgebied, virus-geassocieerde carcinogenese:“Ik zou me kunnen voorstellen dat we de nieuwe High Performance Computing (HPC) zouden kunnen gebruiken om kankerpatiënten of immuungecompromitteerde mensen systematisch te onderzoeken op virussen. weet dat kanker kan worden veroorzaakt door virussen, waarvan het bekendste voorbeeld de menselijke papillomavirussen zijn. Maar waarschijnlijk zien we tot nu toe slechts het topje van de ijsberg menselijk organisme en verhogen het risico op kwaadaardige tumoren."