Wetenschap
Met niet meer dan potlood en papier naar atoomstructuren turen
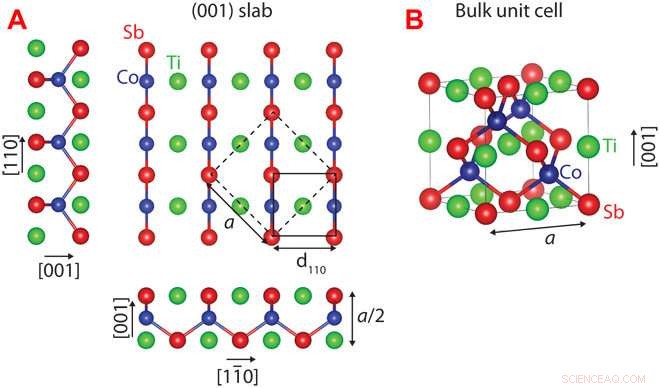
Kristalstructuur van CoTiSb. (A) Unrelaxed (001) plaat van CoTiSb met TiSb-beëindiging. De conventionele bulkeenheidcel is gemarkeerd met stippellijnen (randlengte a), en de (1 × 1) oppervlakte-eenheidscel wordt gemarkeerd door een ononderbroken lijn. (B) Conventionele kubische eenheidscel bestaande uit een CoSb-zinkblende-subrooster dat is gevuld met Ti. Credit: wetenschappelijke vooruitgang (2018). DOI:10.1126/sciadv.aar5832
Wie zou denken dat het ontrafelen van het mysterie van hoe oneindig kleine atomen zich rangschikken aan de randen van kristallen in geavanceerde materialen zo simpel zou kunnen zijn als één, twee, drie?
Het modelleren van de moleculaire structuur van het oppervlak van een kristal vereist normaal gesproken krachtige computers, maar ingenieurs van de University of Wisconsin-Madison hebben een veel eenvoudigere methode bedacht - een methode die net zo eenvoudig is als tellen met potlood en papier.
De simpele strategie zou kunnen helpen om ultrasnelle computerchips tot stand te brengen op basis van andere materialen dan silicium.
"We waren verrast om te ontdekken dat het was, in feite, zo simpel, " zegt Jason Kawasaki, een UW-Madison hoogleraar materiaalkunde en techniek. "Met een paar kleine aanpassingen, we konden structuren voorspellen die kwantitatief zeer nauwkeurig waren."
Ze waren zo nauwkeurig dat zijn nieuwe voorspellingsaanpak, gepubliceerd op 1 juni 2018 in het journaal wetenschappelijke vooruitgang , biedt een snelle en gemakkelijke procedure om in te zoomen op veelbelovende materialen voor gebruik in geavanceerde elektronica zoals kwantumcomputers die problemen veel sneller oplossen dan conventionele op silicium gebaseerde machines.
"Voordat we materialen op interessante manieren kunnen gebruiken voor apparaten van de volgende generatie, je moet begrijpen hoe de structuur aan de oppervlakte verandert, ", zegt Kawasaki.
Het nauwkeurig voorspellen van kristaloppervlakstructuren is een probleem waar wetenschappers al lang mee bezig zijn. Atomen aan de rand van een materiaal hebben de neiging zichzelf te herschikken, verliezen soms hun elektronische of magnetische eigenschappen.
Kawasaki en collega's concentreerden zich op een soort materiaal dat half-Heusler-verbindingen wordt genoemd, die verschillende afstembare elektronische en magnetische eigenschappen hebben. Helaas, veel half-Heuslers presteren niet zoals voorspeld wanneer ze worden gecombineerd met andere materialen of worden teruggebracht tot een plat oppervlak.
"Als je kleine herschikkingen van atomen hebt, u kunt grote veranderingen van eigenschappen hebben, ", zegt Kawasaki.
Alle materialen zijn opgebouwd uit atomen, die kernen in hun centra hebben die worden omringd door steeds veranderende wolken van kleine subatomaire deeltjes die elektronen worden genoemd. Atomen kunnen zich verbinden, of obligatie, door een deel van hun elektronen met elkaar te delen. Kristallen bestaan uit vele atomen die aan elkaar zijn gebonden in een regelmatig gerangschikt en zich herhalend patroon. Dat patroon breekt, echter, op kristaloppervlakken of interfaces, waardoor sommige atomen zonder partners en ongedeelde elektronen aan het bulkmateriaal bungelen.
Binnen het starre interieur van kristallen, geavanceerde simulaties kunnen atomaire arrangementen bepalen, maar computers hebben de eerste beste schattingen van configuraties nodig om structurele voorspellingen te doen.
Voor een lange tijd, de beste gissingen aan de oppervlakte waren onmogelijk omdat de aanwezigheid van bungelende elektronen ervoor zorgt dat het aantal mogelijke conformaties omhoogschiet.
"De juiste tools en het juiste theoretische kader bestonden niet, ", zegt Kawasaki.
Het juiste theoretische kader bleek verrassend eenvoudig, beheerst door elementaire scheikunderegels. Het enige dat nodig is, is het tellen van alle elektronen die elk atoom naar de oppervlakte brengt, tel alle elektronen op waarvan wordt voorspeld dat ze in bindingen zijn, en bepalen of die nummers overeenkomen. Als alle elektronen zijn opgeteld, de structuur is waarschijnlijk stabiel. Als niet, het is terug naar de tekentafel.
Het tellen is zo eenvoudig dat Kawasaki letterlijk potlood en papier kan gebruiken om berekeningen uit te voeren.
Het is bekend dat telregels goed werken voor eenvoudige materialen. Echter, wetenschappers gingen ervan uit dat de elektronenwolken voor de metaalatomen waaruit half-Heusler-materialen bestaan, te ingewikkeld waren voor een dergelijke basisboekhouding.
Kawasaki en collega's bewezen dat idee verkeerd.
"We ontdekten dat veel van de algemene regels die zijn ontwikkeld voor het begrijpen van hechting in eenvoudige systemen, kunnen worden toegewezen aan deze complexere materialen, ", zegt Kawasaki.
Met behulp van deze aanpak, Kawasaki en collega's voorspelden en bevestigden de oppervlakteconfiguratie voor een belangrijk half-Heusler-materiaal, kobalttitaniumantimoon genaamd, wat een potentieel bruikbare halfgeleider is. De onderzoekers maten het kristaloppervlak met geavanceerde beeldvormingstechnieken, en merkten op dat hun potlood-en-papier voorspellingen perfect overeenkwamen met echte atomaire configuraties.
De onderzoekers pasten hun methode vervolgens toe op nog twee half-Heusler-verbindingen, een halfmetaal en een ferromagneet, en ze zijn van plan om meer veelbelovende materialen te identificeren.
Kawasaki voerde de kristalgroei- en meetexperimenten uit in samenwerking met Chris Palmstrøm, een faculteitslid in elektrische en computertechniek en materiaalkunde aan de Universiteit van Californië, Santa Barbara.
 Berekening van de tijd om een object te verwarmen
Berekening van de tijd om een object te verwarmen- Nieuwe genomische methode onthult atomaire rangschikkingen van batterijmateriaal
- Een model van een atoom maken van Styrofoam
- Snel gecontroleerd transport van waterdruppels door door zonlicht aangedreven pomp
- Chemici vinden nieuwe manier om de bouwstenen van veel medicijnen te maken
- Slimme transferregels kunnen EU-klimaatbeleid versterken
- Het noordpoolgebied staat in brand:de Siberische hittegolf alarmeert wetenschappers
- Eerste directe metingen van sedimenten in de Stille Oceaan onthullen sterke methaanbron
- Aardbeving rammelt Indonesische hoofdstad, maar geen schade zichtbaar
- Delhi's gecompliceerde luchtvervuilingsprobleem
Hoofdlijnen
- Sommige planten worden groter - en gemener - als ze worden geknipt, studie vondsten
- Nieuwe statistische methode voor het evalueren van reproduceerbaarheid in studies van genoomorganisatie
- Hoe Punnett Squares
- Genetische ontdekking nog een hulpmiddel in de strijd tegen tarweplagen
- Niveaus van celorganisatie
- Het verschil tussen craniologie en frenologie
Craniologie en frenologie zijn beide praktijken die de conformatie van de menselijke schedel onderzoeken; echter, de twee zijn heel verschillend. Craniologie is de studie van verschillen in vorm, groott
- Structuur van het hart Cell
- Chemische stoffen gebruikt in de forensische wetenschap
- Biologen onderzoeken de moleculaire onderbouwing van cellen die herstellen van de rand van geprogrammeerde dood
- Koolstof nanobuistransistors kunnen leiden tot goedkope, flexibele elektronica

- All-carbon zonnecel maakt gebruik van infrarood licht

- Voorbij 1 en 0:ingenieurs vergroten potentieel voor het creëren van opvolger van krimpende transistors

- Nieuwe analytische technologie onthult nanomechanische oppervlaktekenmerken
- Onderzoekers maken 's werelds grootste DNA-origami


 Microscopische zwemmers met visuele waarneming van groepsleden vormen stabiele zwermen
Microscopische zwemmers met visuele waarneming van groepsleden vormen stabiele zwermen- Hoe maak je een eierdruppelcontainer met rietjes
- Glaselektroden gebruikt in pomp op nanoschaal
- Amazon Go opent tweede kassaloze supermarkt
- Wat betekenen de cijfers in recyclingsymbolen op plastic artikelen?
- Was aan, wegsmelten
- Backcountry skiërs opgelet:wetenschappers willen je hulp
- Foodora trekt zich terug uit Australië
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
