Wetenschap
Team brengt subatomaire resolutie naar computationele microscoop
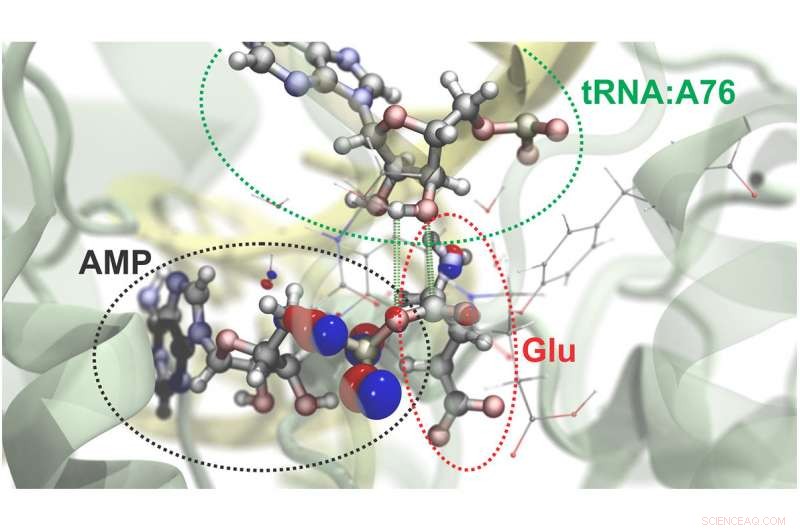
Onderzoekers kunnen atomaire en subatomaire dynamica in grote moleculaire systemen simuleren. Hier is een visualisatie van het proces waarbij het aminozuur glutamaat (Glu) wordt gehecht aan een specifiek gebied van zijn transfer-RNA (tRNA). Een energierijk molecuul, ATP, stuurt deze reactie aan en wordt daarbij omgezet in AMP. De rode en blauwe bubbels vertegenwoordigen de kans op het vinden van elektronen in bepaalde regio's. Groene stippellijnen geven de atomen aan die zich bij deze chemische reactie binden. Krediet:Rafael Bernardi, Zan Luthey-Schulten en Marcelo Melo
Wetenschappers hebben een "computationele microscoop" gebouwd die de atomaire en subatomaire krachten kan simuleren die moleculaire interacties aansturen. Deze tool stroomlijnt de inspanningen om de chemie van het leven te begrijpen, modelleren van grote moleculaire systemen en het ontwikkelen van nieuwe farmaceutische en industriële middelen, zeggen de onderzoekers.
Ze rapporteren hun bevindingen in het tijdschrift Natuurmethoden .
De wetenschappers combineerden twee computationele benaderingen die werden gebruikt om moleculaire interacties te simuleren. De eerste, een moleculaire-dynamicaprogramma op nanoschaal dat bekend staat als NAMD, gebruikt klassiek-mechanische methoden om de structuur te modelleren en het gedrag van honderden miljoenen individuele atomen te simuleren. Het tweede programma zoomt in op het subatomaire rijk, het simuleren van de interacties van protonen, neutronen en elektronen. Modelleren op deze kwantummechanische schaal vereist veel rekenkracht, dus implementeerden de onderzoekers een methode om grote moleculen te verdelen in klassieke en kwantummechanica-regio's. Hierdoor kunnen ze hun computerbronnen concentreren op kleine regio's die betrokken zijn bij kritieke interacties, zoals het maken of verbreken van chemische bindingen.
Zowel moleculaire mechanica als kwantummechanica programma's zijn al jaren beschikbaar, en andere teams hebben gewerkt om ze te combineren, zei professor scheikunde van de Universiteit van Illinois, Zaida (Zan) Luthey-Schulten, die samen met haar man het nieuwe onderzoek leidde, U. of I. natuurkunde professor Klaus Schulten. Maar de nieuwe inspanning stroomlijnt het proces van het opzetten, het uitvoeren en analyseren van de simulaties.
"We hebben het zo opgezet dat onderzoekers gemakkelijk kunnen kiezen hoe ze hun eigen systemen indelen, " zei Luthey-Schulten. "Mijn eigen studenten proberen het uit, en de meesten van hen zijn in staat om het zonder veel moeite te doen."
Schulten ontwikkelde NAMD in Illinois in 1995, combineren met visualisatiesoftware, VMD, waarmee onderzoekers kunnen zien hoe grootschalige moleculaire interacties zich ontvouwen. Schulten, die in 2016 stierf, stelde deze benadering gelijk aan "het bouwen van een computationele microscoop".
De computationele microscoop is ideaal voor het modelleren van structurele eigenschappen en bewegingen van grote complexen. Bijvoorbeeld, in 2013, Schulten en zijn collega's gebruikten NAMD om de hiv-capside te modelleren, die uit meer dan 1 bestaat 300 identieke eiwitten die samenkomen in een kooiachtige structuur die het virus beschermt totdat het een gastheercel binnengaat. Die simulatie was verantwoordelijk voor de interacties van meer dan 64 miljoen atomen en vereiste het gebruik van de Blue Waters-supercomputer in het National Center for Supercomputing Applications aan de U. of I. De nieuwe studie maakte ook gebruik van Blue Waters, dit keer om de resolutie van de computationele microscoop te verbeteren.

Van links, afgestudeerde student Marcelo Melo, hoogleraar scheikunde Zaida Luthey-Schulten, postdoctoraal onderzoeker Rafael Bernardi en hun collega's hebben een nieuwe benadering ontwikkeld voor het modelleren van grote moleculaire interacties op atomaire en subatomaire schaal. Hun werk stroomlijnt de methode voor andere wetenschappers en studenten. Krediet:L. Brian Stauffer
De NAMD-software is ontworpen om het gedrag van individuele atomen te beschrijven. Maar individuele atomen die betrokken zijn bij specifieke chemische interacties en reacties gedragen zich niet altijd zoals hun tegenhangers elders. Om te begrijpen hoe ze variëren, moeten de subatomaire krachten die in het spel zijn nader worden bekeken. Dit is vooral belangrijk in de dynamische gebieden van moleculen - bijvoorbeeld, die plaatsen waar chemische bindingen worden gemaakt of verbroken, aldus de onderzoekers.
In de nieuwe studie het onderzoeksteam in Illinois werkte samen met QM-experts Frank Neese, van het Max Planck Instituut voor kolenonderzoek in Mulheim an der Ruhr, Duitsland; en Gerd B. Rocha, van de Federale Universiteit van Paraiba, in Joaoo Pessoa, Brazilië.
Als demonstratie van de nieuwe aanpak, de onderzoekers simuleerden het chemische gedrag van transfer-RNA's, moleculen die een sleutelrol spelen bij het vertalen van genetische informatie in eiwitten. Met behulp van NAMD, ze hebben de algehele moleculaire structuur van tRNA gemodelleerd op het moment dat een speciaal eiwit een aminozuur in het tRNA laadt. Ze verdeelden twee locaties van het complex in regio's die de meer gerichte kwantummechanische benadering vereisen. (Bekijk een filmpje van de simulatie.)
Dankzij de subatomaire simulaties van de interacties van de twee regio's kon het team simulaties uitvoeren van vier verschillende scenario's waardoor het tRNA zou kunnen functioneren zoals in de cel. Hun simulaties onthulden dat een van de vier potentiële chemische routes energetisch gunstiger was dan de andere en dus waarschijnlijker was.
De onderzoekers gebruikten ook verschillende methoden om het tRNA-complex te verdelen tussen de MM- en QM-regio's en rapporteerden over elke benadering.
"We hebben niet slechts één manier gekozen, we hebben er zoveel mogelijk gekozen. We geven de gebruiker vrijheid. Hoe je het structureert, hangt echt af van het specifieke systeem dat je bestudeert, " zei U. of I. postdoctoraal onderzoeker Rafael Bernardi, een co-hoofdauteur van de studie met afgestudeerde student Marcelo Melo.
"We doen het hele systeem niet kwantummechanisch, want dat zou een eeuwigheid duren om te berekenen, ' zei Melo.
"NAMD is ontworpen - en dit was de visie van mijn man - om echt grote systemen te behandelen, " zei Luthey-Schulten. "Nu kunnen we daar de subatomaire schaal aan toevoegen, het openen van enorme nieuwe mogelijkheden voor onderzoek."
 Voorspellen en beheersen van dioxines
Voorspellen en beheersen van dioxines- NZs-fossielen laten zien dat meer soorten in warmere wateren leefden, maar de huidige opwarmingstrends kunnen dit patroon doorbreken
- Duikers vinden gezonken schat uit het Romeinse tijdperk in scheepswrak bij Israël
- Hoe CEO's, experts en filosofen zien de grootste risico's ter wereld anders
- Afbeelding:voorgeschreven branden om bosbranden in Arizona te voorkomen
Hoofdlijnen
- Nieuw gevonden eiwit kan virale infectie en herpes-geïnduceerde kanker voorkomen
- Het belang van plantaardige cellen
- Nieuw onderzoek naar spermastamcellen heeft gevolgen voor mannelijke onvruchtbaarheid en kanker
- Microbiologie vs. Biochemie
- Hoe zijn fytoplankton reproductie?
- Wanneer is de beste tijd van de dag om een beslissing te nemen?
- Honden en wolven delen parasieten
- 10 echt slimme mensen die echt domme dingen deden
- Waarom is water belangrijk voor levende organismen?
- Nano-beeldvorming van intersubband-overgangen in 2D-materialen met weinig lagen
- Atoomstralen schieten rechter via trapsgewijze siliconen peashooters

- Klassieke synchronisatie duidt op aanhoudende verstrengeling in geïsoleerde kwantumsystemen

- Add-on clip maakt van smartphone een volledig operationele microscoop

- De toekomst van herschrijfbaar geheugen schrijven

 Gevaar niet voorbij:gaslekken, schimmel weefgetouw voor Harvey evacués
Gevaar niet voorbij:gaslekken, schimmel weefgetouw voor Harvey evacués- Broeikasgasanalyses maken op de weg, eh, rails
- Welke planten leven in de Atlantische Oceaan?
- Afbeelding:Hubbles-plakje Boogschutter
- Wat is metallurgische cokes?
- Hoe de massa van een proton te berekenen
- Hoe leveranciers van alledaagse apparaten u kwetsbaar maken voor cyberaanvallen - en wat u eraan kunt doen
- Jonge boeren in Californië vinden nieuwe manieren om vee te fokken en het land te verbeteren
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
