Wetenschap
Het versnellen van geometrie-optimalisatie in moleculaire simulatie
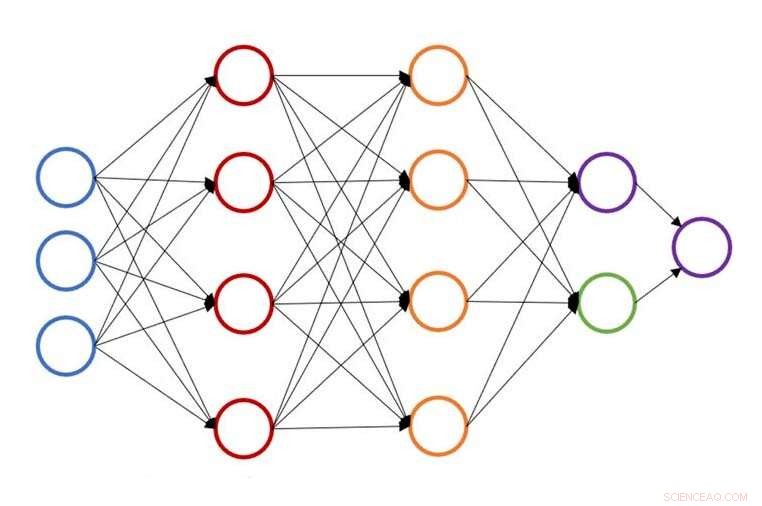
Illustratie van een enkele neurale netwerkstructuur voor voorspelling van atoomenergie. Krediet:John Kitchin, Carnegie Mellon Universiteit
machinaal leren, een data-analysemethode die wordt gebruikt om het bouwen van analytische modellen te automatiseren, heeft de manier waarop wetenschappers en ingenieurs onderzoek doen hervormd. Een tak van kunstmatige intelligentie (AI) en informatica, de methode steunt op een groot aantal algoritmen en brede datasets om patronen te identificeren en belangrijke onderzoeksbeslissingen te nemen.
Er ontstaan toepassingen van machine learning-technieken op het gebied van oppervlaktekatalyse, uitgebreidere simulaties van nanodeeltjes mogelijk maken, studies over segregatie, structuur optimalisatie, on-the-fly leren van krachtvelden, en screening met hoge doorvoer. Echter, het verwerken van grote hoeveelheden gegevens kan vaak een lange en rekenkundige taak zijn.
Geometrie optimalisatie, vaak de snelheidsbeperkende stap in moleculaire simulaties, is een belangrijk onderdeel van computationele materialen en oppervlaktewetenschap. Het stelt onderzoekers in staat om atomaire structuren en reactiepaden in de grondtoestand te vinden, eigenschappen die worden gebruikt om de kinetische en thermodynamische eigenschappen van moleculaire en kristalstructuren te schatten. Hoewel van vitaal belang, het proces kan relatief langzaam zijn, waarvoor een groot aantal berekeningen nodig is.
Aan de Carnegie Mellon-universiteit, John Kitchin werkt eraan om dat proces te versnellen door een op neurale netwerken gebaseerde actieve leermethode te bieden die geometrische optimalisatie voor meerdere configuraties tegelijk versnelt. Het nieuwe model verlaagt het aantal dichtheidsfunctionaaltheorie (DFT) of effectieve mediumtheorie (EMT) berekeningen met 50 tot 90 procent, waardoor onderzoekers hetzelfde werk in minder tijd of meer werk in dezelfde hoeveelheid tijd kunnen doen.
"Normaal gesproken, wanneer we geometrie-optimalisatie doen, we beginnen vanaf nul, "zei Kitchin. "De berekeningen profiteren zelden van iets wat we in het verleden wisten."
"Door een surrogaatmodel aan het proces toe te voegen, we hebben het mogelijk gemaakt om te vertrouwen op eerdere berekeningen, in plaats van elke keer opnieuw te beginnen."
De studie illustreert de versnelling van verschillende casestudies, inclusief oppervlakken met adsorbaten, kale metalen oppervlakken, en stootte elastische band voor twee reacties. In ieder geval, het Atomic Simulation Environment (ASE)-optimizer Python-pakket maakte minder DFT-berekeningen mogelijk dan de standaardmethode.
Het ASE-optimizer Python-pakket is beschikbaar gesteld aan collega-ingenieurs en wetenschappers om het gebruik van neuraal netwerkensemble actief leren voor geometrie-optimalisatie gemakkelijker te maken.
 Science Experiment Ideas: Epsom Salts
Science Experiment Ideas: Epsom Salts - Spierachtig materiaal zet uit en trekt samen als reactie op licht
- Een eeuwenoud model voor het ontstaan van levens krijgt flinke onderbouwing
- Kunnen bacteriën sterkere auto's maken, vliegtuigen en bepantsering?
- Kruiskoppelingsreacties:semiheterogene op PCN-Cu gebaseerde metallafotokatalyse
Hoofdlijnen
- Is de computer een goed model voor de hersenen?
- De wetenschap ontdekt waarom sommigen van ASMR-video's houden en anderen ze haten
- Meer dan een getallenspel:nieuwe techniek meet microbiële gemeenschappen op biomassa
- Zorgen voor het voortbestaan van olifanten in Laos:een kwestie van economie
- Inspanningen om zeeschildpadden te redden zijn een wereldwijd succesverhaal op het gebied van natuurbehoud:wetenschappers
- De effecten van straling op dieren
- De rol van de longen
- Wat is ongelijk verdeeld in de vrouwelijke cytokinese?
- Voorbeelden van stoffen die gefaciliteerde diffusie gebruiken
- Kunstmatig-intelligentiesysteem ontwerpt medicijnen helemaal opnieuw
- Faseovergangsdynamiek in tweedimensionale materialen

- Ooggestuurde zachte lens maakt de weg vrij voor zachte mens-machine-interfaces
- Hoe natriumcarbonaatoplossing te maken

- Onderzoekers vinden een nieuw verband tussen celmetabolisme en celdeling

 Voor platinakatalysatoren, kleiner is misschien beter
Voor platinakatalysatoren, kleiner is misschien beter- Fopspeenbiosensor kan helpen bij het bewaken van de gezondheid van pasgeborenen
- De oude Mongoolse schedel is de vroegste moderne mens die tot nu toe in de regio is gevonden
- ESA-satelliet rijdt vanuit Rusland in een baan om de aarde
- MPH omzetten in voet per seconde
- CubeSats in beweging krijgen:M-Argo zal de eerste zijn die de interplanetaire ruimte op eigen kracht doorkruist
- Om een bloeiende cafécultuur in stand te houden, we moeten de wegwerpbeker weggooien.
- Modellering toont dringende noodzaak om de aanwerving en arbeidsvoorwaarden voor astronomen te vernieuwen
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
