Wetenschap
Structuurmotiefgericht leerkader voor anorganische kristallijne systemen
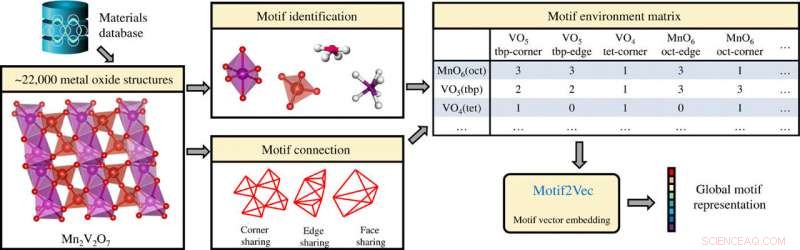
Extractie van structuurmotiefinformatie in anorganische kristallijne verbindingen (metaaloxiden) en het genereren van globale motiefrepresentaties met behulp van de motiefomgevingsmatrix. Credit: wetenschappelijke vooruitgang , doi:10.1126/sciadv.abf1754
Fysieke principes kunnen worden opgenomen in een machine learning-architectuur als een fundamentele opzet om kunstmatige intelligentie voor anorganische materialen te ontwikkelen. In een nieuw verslag nu op wetenschappelijke vooruitgang , Huta R. Banjade, en een onderzoeksteam in de natuurkunde, computer- en informatiewetenschap en nanowetenschap in de VS en België stelden structuurmotieven in anorganische kristallen voor om als centrale input te dienen voor een raamwerk voor machinaal leren. Het team demonstreerde hoe de aanwezigheid van structuurmotieven en hun verbindingen in een groot aantal kristallijne verbindingen kon worden omgezet in unieke vectorrepresentaties via een niet-gecontroleerd leeralgoritme. Ze bereikten dit door een motiefgericht leunend raamwerk te creëren door motiefinformatie te combineren met op atomen gebaseerde neurale netwerken om een atoom-motief dubbel graafnetwerk (AMDNet) te vormen. De opstelling voorspelde nauwkeurig de elektronische structuur van metaaloxiden zoals bandgaps. Het werk illustreert een methode om neurale netwerkleerarchitecturen te ontwerpen om complexe materialen te onderzoeken die verder gaan dan de fysieke eigenschappen van atomen.
ML-methoden
Methoden voor machinaal leren (ML) kunnen worden gecombineerd met enorme materiaalgegevens om de ontdekking en het rationele ontwerp van functionele vaste-stofverbindingen te versnellen. Gesuperviseerd leren kan leiden tot voorspellingen van materiële eigenschappen, inclusief fasestabiliteit en kristalaard, effectief voor simulaties van molecuuldynamica. Structuurmotieven kunnen worden gemaakt in overeenstemming met de eerste regel van Pauling, door een gecoördineerd veelvlak van anionen te vormen rond elk kation in een verbinding om zich te gedragen als fundamentele bouwstenen die sterk gecorreleerd zijn met materiaaleigenschappen. Bijvoorbeeld, de structuurmotieven in kristallijne verbindingen kunnen een essentiële rol spelen bij het bepalen van de materiaaleigenschappen in verschillende technische en wetenschappelijke toepassingen. In dit werk, Banjade et al. structuurmotiefinformatie opgenomen in een machineleunend (ML) raamwerk. De wetenschappers combineerden de motiefinformatie met grafische convolutionele neurale netwerken om een op motief gerichte deep learning-architectuur te ontwikkelen die bekend staat als atom-motif dual graph network (AMDNet). De nauwkeurigheid van de structuur overtrof die van een bestaand state-of-the-art op atomen gebaseerd grafieknetwerk om de elektronische structuren van anorganische kristallijne materialen te voorspellen.
De t-gedistribueerde stochastische buur-inbeddingsprojectie van motiefvectoren geconstrueerd met behulp van de motiefomgevingsmatrix. De motiefclusters 1 t/m 4 zijn gekoppeld aan verschillende motieftypen, waaronder (1) kubus, (2) kuboctaëder, (3) octaëder, en (4) een mengsel van tetraëder (in magenta) en vierkant vlak (in overblijfsel). t-SNE, t-gedistribueerde stochastische inbedding van buren. Credit: wetenschappelijke vooruitgang , doi:10.1126/sciadv.abf1754 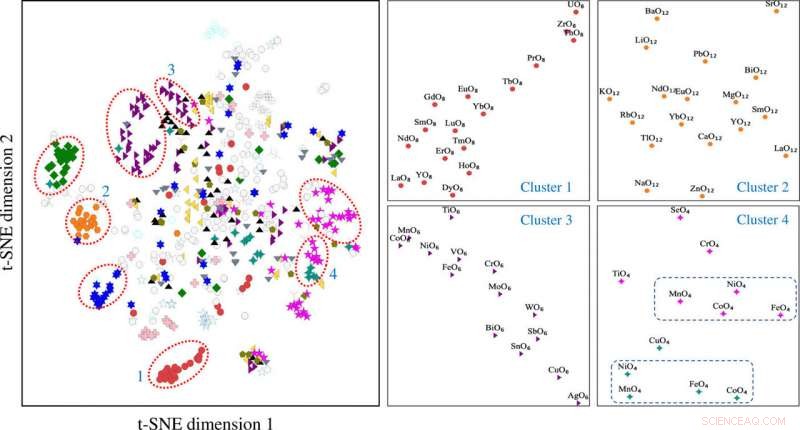
Een niet-gesuperviseerd leeralgoritme Atom2Vec kan hoogdimensionale vectorrepresentaties van atomen begrijpen door basiseigenschappen van atomen te coderen op basis van een uitgebreide database met chemische formules. Banjade et al. gericht op binaire en ternaire metaaloxiden die een enorme en diverse materiële ruimte vormen waar kristalstructuren worden gekarakteriseerd via kation-zuurstofcoördinatie. Om de informatie over het structuurmotief te extraheren, het team gebruikte de lokale omgevingsidentificatiemethode ontwikkeld door Waroquiers et al. zoals geïmplementeerd door de Pymatgen-code. Het team identificeerde drie verschillende soorten connectiviteit tussen een motief en het aangrenzende motief; inclusief innerlijk delen (één atoom gedeeld), edge sharing (twee atomen gedeeld), en gezicht delen (drie of meer atomen gedeeld). De wetenschappers stelden vervolgens een leeralgoritme voor om te profiteren van het motiefgegevensverzamelingsproces en converteerden effectief elke rij van de motiefomgevingsmatrix in een hoogdimensionale vector om een uniek structuurmotief weer te geven. Vervolgens extraheren ze motiefinformatie voor het leerproces met behulp van een grafisch convolutienetwerk. Het team wilde patronen en clusterinformatie voor deze hoogdimensionale motiefvectoren identificeren om de complexe materiaaleigenschappen van oxideverbindingen te beïnvloeden. Ze visualiseerden de hoogdimensionale gegevens met behulp van de t-gedistribueerde stochastische inbedding (t-SNE) - een niet-lineaire dimensionaliteitsreductietechniek.
Motiefinformatie gebruiken in neurale netwerken in grafieken.
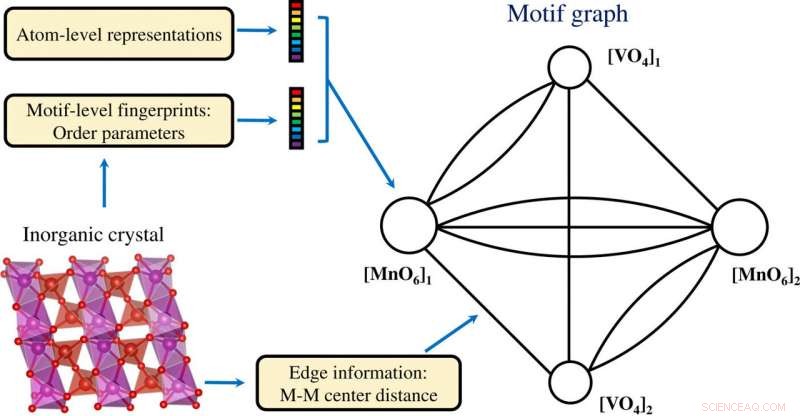
Constructie van een motiefgrafiek op basis van informatie op zowel atoomniveau als motiefniveau gecodeerd in een anorganisch kristal. Credit: wetenschappelijke vooruitgang , doi:10.1126/sciadv.abf1754
De wetenschappers verkregen geprojecteerde motiefvectorgegevens in twee dimensies met behulp van het t-SNE-proces. Ze merkten verschillende clusters op op basis van de motieftypen. De chemische eigenschappen van de elementen die de motieven vormen, speelden een sleutelrol tijdens de clustervorming. Bijvoorbeeld, Op lanthanide gebaseerde motieven vormden verschillende clusters op basis van motieftype en op yttrium gebaseerde motieven bleven dicht bij de op lanthanide gebaseerde motieven vanwege hun chemische overeenkomsten. Motieven geassocieerd met zink en magnesium kwamen ook samen. De niet-gecontroleerde, op leren gebaseerde bevindingen ondersteunden de structuurmotieven om te dienen als essentiële input voor kristallijne verbindingen die elementaire en structurele informatie dragen. Het team gebruikte vervolgens structuurmotiefinformatie als essentiële input voor een graaf-neuraal netwerk (GNN) om fysieke eigenschappen van materialen te voorspellen. De meeste grafennetwerken waren van toepassing op kristallijne materialen. Om een leerarchitectuur van atoom- en motief-niveau grafiekrepresentaties van materialen mogelijk te maken, Banjade et al. stelde voor dat AMDNet zou kunnen worden geconstrueerd om het leerproces te verbeteren en de voorspellingsnauwkeurigheid voor de elektronische structuureigenschappen van metaaloxiden te verbeteren. In de motiefgrafieken de onderzoekers codeerden informatie op atoomniveau en op motiefniveau in elk knooppunt en construeerden de motiefgrafiek, inclusief uitgebreide connectiviteit, hoek, afstand en volgorde parameters met behulp van Python pakket robokristallografie.
AMDNet
In de voorgestelde AMDNet-architectuur, Banjade et al. motiefinformatie opgenomen in een leerraamwerk voor graafnetwerken om motiefgrafieken en atoomgrafieken te genereren die verbindingen met verschillende kardinaliteit van randen en knooppunten vertegenwoordigen om de informatie te combineren voordat voorspellingen worden gedaan. Voor elk materiaal, het team genereerde een atoomgrafiek en een motiefgrafiek. Ze gebruikten toen 22, 606 binaire en ternaire metaaloxiden uit de Materials Project-database om de effectiviteit van het voorgestelde model te testen en gericht op de voorspelling van bandgaps - een complex elektronisch structuurprobleem. De resultaten toonden de superioriteit van AMDNet tijdens bandgap-voorspelling in vergelijking met voorgaande netwerken. Het model toonde ook superieure prestaties tijdens een classificatietaak van metaal versus niet-metaal. Het werk toonde de eerste inspanningen om materiële informatie op hoog niveau op te nemen in deep learning-modellen voor solid-state materialen.
AMDNet-architectuur en voorspellingen van materiaaleigenschappen. (A) Demonstratie van de leerarchitectuur van het voorgestelde atom-motief dual graph-netwerk (AMDNet) voor het effectief leren van elektronische structuren en andere materiaaleigenschappen van anorganische kristallijne materialen. (B) Vergelijking van voorspelde en werkelijke bandgaps [van berekeningen van de dichtheidsfunctionaaltheorie (DFT)] en (C) vergelijking van voorspelde en werkelijke vormingsenergieën (van DFT-berekeningen) in de testdataset met 4515 verbindingen. Credit: wetenschappelijke vooruitgang , doi:10.1126/sciadv.abf1754 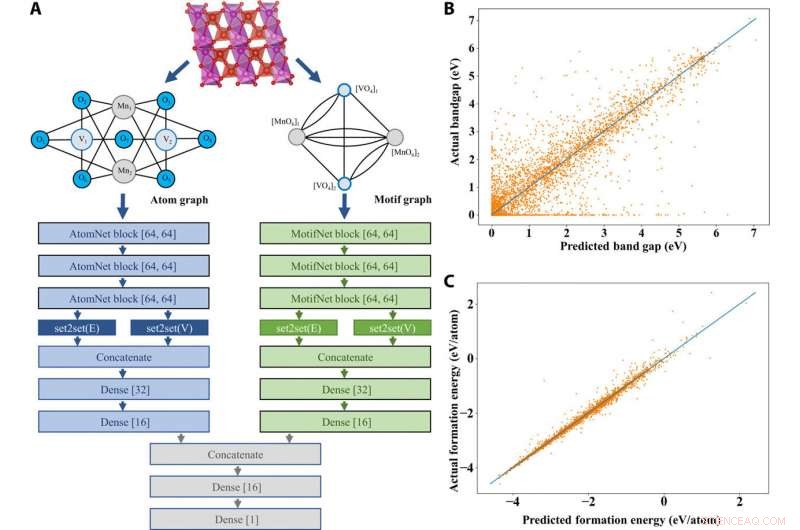
Op deze manier, Huta R. Banjade en collega's lieten zien hoe structuurmotieven in kristalstructuren kunnen worden gecombineerd met niet-gecontroleerde en gesuperviseerde machinale leermethoden om de effectieve weergave van solid-state materiaalsystemen te verbeteren. Voor complexe elektronische structuren, het team heeft de structuur- en motiefverbindingsinformatie opgenomen in een AMDNet-model om beter te presteren dan bestaande netwerken en om de elektronische bandgaps en classificatietaken tussen metaal en niet-metaal te voorspellen. Dit algemene leerraamwerk kan worden gebruikt om andere materiaaleigenschappen te voorspellen, waaronder mechanische en aangeslagen toestandseigenschappen in tweedimensionale materialen en metaal-organische raamwerken.
© 2021 Science X Network
 Wetenschappers ontwerpen materiaal dat energie kan opslaan als een adelaarsgreep
Wetenschappers ontwerpen materiaal dat energie kan opslaan als een adelaarsgreep- Beton met verbeterd slagvastheid voor verdedigingsconstructies
- Actief zeven kan dialyse- en waterzuiveringsfilters verbeteren
- Ontdekking kan game-changer zijn voor farmaceutische producten
- Twee eenvoudige bouwstenen produceren complex 3D-materiaal
- Gaza pompt afvalwater rechtstreeks in zee naarmate de crisis verergert
- Geliefde 600 jaar oude witte eik maakt laatste buiging
- NASA vindt weinig verbetering in Miriams structuur
- NASA-satellieten kijken in een scheve orkaan Maria
- Maleisië dringt bij EU aan op gepland verbod op palmolie in biobrandstoffen
Hoofdlijnen
- De functie van NIMA-gerelateerd kinase 6 in de rechte groei van plantencellen
- Hoe helpt marihuana de gezondheid van mensen
- Op feiten gebaseerde tips om uw geheugen te verbeteren
- Een geslacht van Europese papierwespen voor het eerst herzien met behulp van integratieve taxonomie
- Het verschil tussen het somatische en autonome systeem
- Garnalenvisserij in New England voor minstens een jaar gesloten
- Veren hebben hun eigen geuren, en roofdieren weten het
- Zeldzame vliegende vossen neergeschoten bij gruwelijke aanval in Australië
- Verschillende soorten cellulaire communicatie
- Krachtige röntgenstralen ontsluiten geheimen van kristalvorming op nanoschaal

- Test voor het verminderen van suikers

- Chemici melden biohernieuwbaar, biologisch afbreekbaar plastic alternatief

- Machine-learning onderzoek ontsluit energiebesparende moleculaire kooien

- Matras ontvlambaarheid standaard is een redder in nood, NIST rapport vondsten


 Rusland verwerpt claim ruimtewapens als propaganda
Rusland verwerpt claim ruimtewapens als propaganda- De observaties van de Parkes-radiotelescoop werpen meer licht op het fenomeen van modusomschakeling in PSR J0614+2229
- Nobelprijswinnaar Jody Williams voert campagne tegen moordende robots
- Gebouwen energie geven
- Bio-ingenieurs zetten stap in de richting van een patch die een gebroken hart kan herstellen
- Verschillen tussen een besloten aquifer en een onbegrensde watervoerende laag
- De helling van een tangens berekenen
- Onderzoek naar het idee dat meer uren studeren garant staat voor een hogere onderwijskwaliteit
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
