Wetenschap
Fujitsu ontwikkelt moleculaire simulatietechnologie om effectief nieuwe kandidaat-geneesmiddelen te creëren

Figuur 1:Tweevlakshoek (de hoek gevormd door het vlak gecreëerd door atomen A, B, en C, en het vlak gecreëerd door atomen B, C, en D). Krediet:Fujitsu
Fujitsu Laboratories heeft vandaag de ontwikkeling aangekondigd van moleculaire simulatietechnologie voor het ontdekken van geneesmiddelen die de bindingsaffiniteit nauwkeurig kan inschatten, die staat voor de mate waarin eiwitten die ziekten kunnen veroorzaken (doelwiteiwitten) zich binden aan chemische stoffen die kandidaat-geneesmiddelen kunnen worden. In het proces van medicijnontdekking, er is vraag naar nauwkeurige voorspelling van de bindingsaffiniteit tussen doeleiwitten en chemische stoffen, die een ruwe schatting geeft van de werkzaamheid van een geneesmiddel. Moleculaire simulatietechnologie is in het verleden veel gebruikt als een methode om bindingsaffiniteit te voorspellen, berekenen van de geschatte krachten die optreden tussen atomen in moleculen met behulp van Newtoniaanse mechanica. Het probleem met deze methode, echter, blijft dat de lage mate van nauwkeurigheid van de schatting van de belangrijkste parameters - de mate van torsie op de bindingsplaatsen. Dit betekent dat de nauwkeurigheid van de schatting van de algehele bindingsaffiniteit ook slecht is.
Nutsvoorzieningen, Fujitsu Laboratories heeft moleculaire simulatietechnologie ontwikkeld die de mate van torsie in een chemische stof schat, die direct is verbonden met de voorspelde bindingsaffiniteit. De nieuwe technologie houdt niet alleen rekening met de bindingsplaats waar de torsie zal optreden, maar ook de impact van naburige atomen. Fujitsu Laboratories evalueerde deze technologie voor 190 soorten chemische stoffen, het vergelijken van de resultaten met de juiste resultaten die zijn verkregen bij de berekening van de eerste beginselen en vervolgens het evalueren van het foutenpercentage. Door dit te doen, het kon bevestigen dat het foutenpercentage in de schatting van de mate van torsie was, gemiddeld, een tiende van die van de vorige technologie. Verwacht wordt dat het gebruik van deze nieuwe technologie in op IT gebaseerde geneesmiddelenontdekking, met zijn vermogen om de bindingsaffiniteit van gerichte eiwitten en chemische stoffen nauwkeurig in te schatten, biedt het potentieel voor baanbrekende nieuwe inspanningen op het gebied van de ontdekking van geneesmiddelen die met eerdere benaderingen niet konden worden bereikt.
De ontdekking van nieuwe medicijnen vereist aanzienlijke kosten en tijdsbestekken die in tientallen jaren kunnen worden gemeten, leidend tot een wereldwijde zoektocht naar nieuwe methoden om drugs te ontdekken. Een van de methoden die veel belangstelling heeft gekregen, is op IT gebaseerde geneesmiddelenontdekking, een nieuwe methode voor het ontdekken van medicijnen met behulp van computers die het mogelijk maakt om chemische stoffen te creëren als kandidaten voor nieuwe medicijnen met een hoge kans op succes. Op IT gebaseerde geneesmiddelenontdekking is een centraal punt geworden voor verwachtingen als een baanbrekende technologie voor de creatie van nieuwe geneesmiddelen, omdat, in tegenstelling tot eerdere methoden van vallen en opstaan, waarin chemische stoffen herhaaldelijk worden gemaakt en getest, deze aanpak maakt het mogelijk om chemische stoffen virtueel te ontwerpen en hun effecten in te schatten.
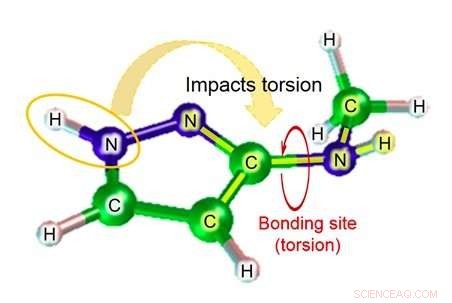
Figuur 2:Voorbeeld van moleculaire structuur:3-(methylamino)pyrazool. Krediet:Fujitsu
De effecten van een chemische stof als medicijn komen tot uiting wanneer de chemische stof zich bindt aan een doeleiwit. Wanneer de chemische stof bindt aan het doeleiwit, het kan zijn vorm veranderen in lijn met die van het doeleiwit. De mate van vervorming, namelijk, de parameters die de omvang van deze vormverandering aangeven, is direct verbonden met de bindingsaffiniteit van de stof en het eiwit, en geeft een globaal beeld van het effect ervan als medicijn. Dit gegeven, er is een sterke vraag naar het vermogen om deze waarde nauwkeurig te voorspellen. Om de mate van vervorming van een chemische stof te berekenen, er zijn methoden gebaseerd op kwantummechanica en methoden gebaseerd op Newtoniaanse mechanica. Op kwantummechanica gebaseerde eerste principes berekening maakt uiterst nauwkeurige berekeningen mogelijk, het oplossen van de toestanden van elektronen uit de typen en posities van de betrokken atomen. Anderzijds, echter, het vermogen van de eerste principes om veeleisende berekeningen uit te voeren, leidt noodzakelijkerwijs tot enorme tijd die nodig is om de berekeningen te voltooien. Om de mate van vervorming voor tal van chemische stoffen te simuleren, benodigde tijd is in de orde van jaren, waardoor deze methode onpraktisch wordt. Anderzijds, geschatte berekeningen op basis van moleculaire simulaties zijn extreem snel, Newtoniaanse mechanica gebruiken om de krachten tussen de atomen in de moleculen te berekenen, en kan zelfs vrij gemakkelijk grote moleculen zoals eiwitten aan. Bijgevolg, deze methode wordt veel gebruikt. Met Newtoniaanse mechanica, de krachten tussen de atomen worden op de volgende manier uitgedrukt:
- Als een kracht die afhangt van de afstand tussen twee aan elkaar gebonden atomen
- Een kracht die afhankelijk is van de hoeken tussen drie aan elkaar gebonden atomen
- Een kracht die afhankelijk is van de mate van torsie in de binding, en
- Een kracht die afhankelijk is van de afstand tussen atomen die niet gebonden zijn.
Tussen deze, wanneer een chemische stof is gebonden aan een doeleiwit, de mate van torsie van de binding vertegenwoordigt de belangrijke mate van vervorming. Met bestaande technologie, echter, de nauwkeurigheid van de schatting van de tweevlakshoek (figuur 1) parameter, die nodig is om de mate van torsie van de binding te berekenen, is vrij laag, resulterend in het probleem van lage nauwkeurigheid bij de schatting van de affiniteit van de binding in de simulatie.
Fujitsu Laboratories ontwikkelt al meer dan tien jaar moleculaire simulatietechnologie. Nutsvoorzieningen, met behulp van de kennis die het heeft verkregen door eerdere inspanningen, Fujitsu Laboratories heeft een moleculaire simulatietechnologie ontwikkeld die de tweevlakshoekparameter kan schatten door rekening te houden met de impact van atomen in de buurt van de binding. Bestaande technologie schat de tweevlakshoekparameter op basis van in totaal vier atomen - de twee atomen in de relevante binding, en de andere atomen waaraan elk van die atomen was gebonden. Afhankelijk van de structuur van het molecuul, echter, er zijn gevallen waarin atomen buiten die vier een aanzienlijke impact kunnen hebben, en in die gevallen de foutenmarge van de schatting kan behoorlijk groot zijn. Met deze technologie, Fujitsu Laboratories heeft een database gemaakt met schattingsformules voor partiële structuurpatronen waarbij de impact van atomen verder weg van de bindingsplaats aanzienlijk zou kunnen zijn, evenals voor de mate van torsie van chemische stoffen die in dat geval zou worden verwacht. Met behulp van de relevante schattingsformule om de mate van torsie te vinden (figuur 2) in het geval van moleculen die overeenkomen met de database voor deelstructuren, het is mogelijk geworden om zelfs zeer nauwkeurige schattingen te maken voor moleculaire torsie, die voorheen moeilijk nauwkeurig te berekenen was.
Toen Fujitsu Laboratories deze technologie integreerde in de software die het had ontwikkeld voor het genereren van geavanceerde parameters voor de krachten tussen atomen (FF-FOM), het was in staat om te bevestigen dat de resultaten overeenkwamen met nauwkeurige berekeningen.

Figuur 3:Evaluatie van de prestaties van tweevlakshoekparameterwaarden met behulp van 190 soorten chemische samengestelde structuren. Krediet:Fujitsu
Toen Fujitsu Laboratories het verschil evalueerde tussen de resultaten van deze technologie en de resultaten van een berekening uit eerste principes voor de schatting van de mate van torsie met 190 soorten chemische stoffen, het was minder dan een tiende van die van de vorige technologie, gemiddeld, 0.6 kcal/mol onder kamertemperatuur thermische schommelingen, bevestigen dat de nieuwe technologie praktisch is. Omdat het de bindingsaffiniteit van doeleiwitten en chemische stoffen nauwkeurig kan schatten, naar verwachting zal het gebruik van deze technologie leiden tot de creatie van baanbrekende nieuwe medicijnen door het gebruik ervan in op IT gebaseerde medicijnontdekking.
 Wat is de standaardmethode voor kalibratie van een geleidbaarheidsmeter?
Wat is de standaardmethode voor kalibratie van een geleidbaarheidsmeter? - Insuline sorteren en afscheiden op vervaldatum
- Een zeldzame prestatie:materiaal beschermt tegen zowel biologische als chemische bedreigingen
- De subtiele rol van oppervlakken bij kleverigheid van ionen
- Bio-geïnspireerde materialen van paardenbloemen
- EU-klimaatwetgeving een stap in de goede richting onderzoekers zeggen:
- Onderzoekers stellen noodzakelijke correcties voor in het mondiale biodiversiteitsbeleid
- Dragonfly leeractiviteiten voor Preschool
- Kooldioxideniveaus bereikten 50% hoger dan pre-industriële tijd
- Kenmerken van de zes koninkrijken van organismen
Hoofdlijnen
- Wat is de functie van aerobe ademhaling?
- Milieubeheer van inheemse naties bij het aanpakken van invasieve soorten
- Noord-Amerikaanse primeur:onderzoekers publiceren wetenschappelijke studie over cannabisproductie
- Hoe werkt het plasmamembraan Homeostasis?
- Wat is het pad van het licht door het oog?
- Verbergen of opgegeten worden, urinechemicaliën vertellen modderkrabben
- Hoe slaaplabs werken
- Deal verbiedt val in Colorado, bureau zegt dat het toch niet wordt gebruikt (update)
- Mannetjes passen de snelheid van het sperma snel aan om rivalen te verslaan, studie vondsten




 Onderzoek toont aan hoe vrouwelijke immuuncellen hun tweede X-chromosoom uitgeschakeld houden
Onderzoek toont aan hoe vrouwelijke immuuncellen hun tweede X-chromosoom uitgeschakeld houden- Hoe levenscyclusanalyses (mis)gebruikt kunnen worden om meer plastic verpakkingen voor eenmalig gebruik te rechtvaardigen
- Italiaanse zeelieden kenden Amerika 150 jaar voor Christoffel Columbus, nieuwe analyse van oude documenten suggereert:
- Smeltend ijsveld in Noorwegen onthult grote verzameling oude pijlen
- Facebook-gegevensverzameling - wat u moet weten
- Dark Energy Survey voltooit missie van zes jaar
- Big data spelen grotere rol nu luchtvaartmaatschappijen service personaliseren
- Potato & Battery Science Projects
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
