Wetenschap
Nieuwe bevindingen kunnen leiden tot goedkopere zonnecellen
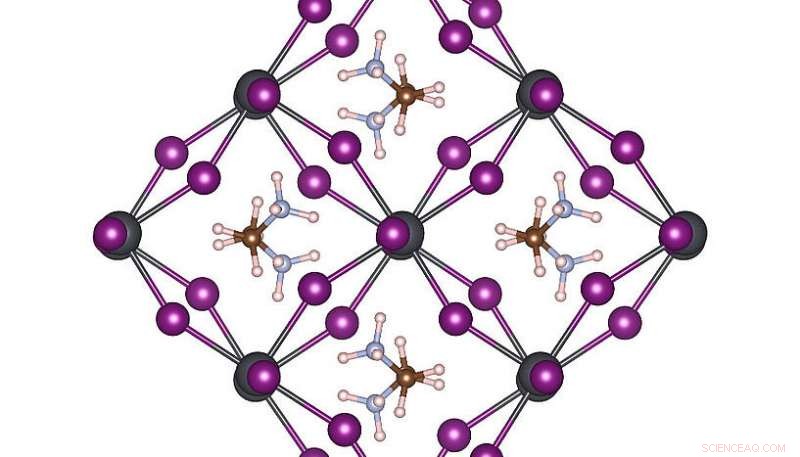
Hoge symmetrie atomaire structuur van MAPbI3 bij kamertemperatuur. Credit:Menno Bokdam/Universiteit van Wenen
Op atomaire schaal kunnen materialen een rijk palet aan dynamisch gedrag vertonen, die direct de fysieke eigenschappen van deze materialen beïnvloedt. Voor vele jaren, het was een droom om deze dynamiek in complexe materialen bij verschillende temperaturen te beschrijven met behulp van computersimulaties. Natuurkundigen van de Universiteit van Wenen hebben een on-the-fly machine learning-methode ontwikkeld die dergelijke berekeningen mogelijk maakt door directe integratie in het op kwantummechanica gebaseerde Vienna Ab-initio Simulation Package (VASP). De veelzijdigheid van de zelflerende methode wordt aangetoond door nieuwe bevindingen, gepubliceerd in het tijdschrift Fysieke beoordelingsbrieven , op de faseovergangen van hybride perovskieten. Deze perovskieten zijn van groot wetenschappelijk belang vanwege hun potentieel voor het oogsten van zonne-energie en andere toepassingen.
Op kamertemperatuur, alle materialen zijn constant in beweging op atomaire schaal. Zelfs massief gesteente bestaat uit rondzwaaiende atomen. De fysieke eigenschappen van materialen zijn direct gekoppeld aan de rangschikking van atomen in de, zogenaamd, kristal rooster. Afhankelijk van de temperatuur of druk kan deze opstelling veranderen, waardoor de materiaaleigenschappen worden beïnvloed. Men kan denken aan diamant, die transparant en hard is vanwege de periodieke rangschikking van koolstofatomen in het diamantkristal. dezelfde atomen, anders geregeld, resulteert in zwart, broos grafiet. Het was al mogelijk om de coördinaten van de atomen nauwkeurig te berekenen in eenvoudige materialen bij verschillende temperaturen met simulaties van kwantummechanische moleculaire dynamica (MD). Echter, dergelijke berekeningen zijn rekenkundig duur en beperken praktische toepassingen tot enkele honderden atomen en beperkte simulatietijd.
Natuurkundigen van de groep Computational Materials Physics aan de Universiteit van Wenen hebben een nieuwe aanpak ontwikkeld die deze beperkingen overwint en simulaties van complexe materialen voor toekomstige energietoepassingen mogelijk maakt. Dit wordt bereikt door een efficiënt en robuust datagestuurd zelflerend algoritme te ontwikkelen en, het belangrijkste, door dit algoritme rechtstreeks te integreren in het Vienna Ab-initio Simulation Package (VASP). Bij de nieuwe aanpak de "machine" kan ophalen, op zichzelf, de essentiële ingrediënten voor een eenvoudigere modelbeschrijving van de op elkaar inwerkende atomen tijdens MD-simulaties. Al na het berekenen van enkele honderden tijdstappen kan de machine de posities van de atomen in de opeenvolgende tijdstappen nauwkeurig genoeg voorspellen. De machine kan ook een schatting maken van de nauwkeurigheid voor de opeenvolgende stappen. Als de fout te groot is, de machine schakelt en voert de zeer nauwkeurige, maar duur, MD berekeningen. Hoe meer simulatietijd verstrijkt, hoe meer de machine leert en hoe nauwkeuriger hij wordt. Op deze manier, er zijn steeds minder MD-berekeningen nodig, wat er uiteindelijk toe leidt dat alle tijdstappen door de machine worden gemaakt. Bovendien, het on-the-fly zelflerend vermogen vermindert de behoefte aan menselijke tussenkomst die vereist is door andere bestaande machine-leermethoden.
Om de kracht van deze nieuwe methode te demonstreren, de onderzoekers hebben het toegepast om de overgangen tussen verschillende atomaire structuren van de MAPbI . te bestuderen 3 perovskiet bij het veranderen van de temperatuur. Dit materiaal is erg populair vanwege zijn potentieel als nieuwe goedkope zonnecelcomponent. Het is gemaakt van organische moleculen die snel kunnen ronddraaien, van elkaar gescheiden door een rooster bestaande uit lood- en jodide-atomen. Afhankelijk van de temperatuur worden drie verschillende kristalfasen gevormd. De atomaire mechanismen in de buurt van de overgangstemperatuur zijn erg moeilijk experimenteel te bepalen, en MD-simulaties zouden zelfs op een modern supercomputersysteem jaren rekentijd vergen. Na het leren, de machine kan de faseovergangstemperaturen en roosterconstanten van dit materiaal met ongekende precisie voorspellen. De ontwikkelde methode is algemeen en toepasbaar op vele andere toekomstige materiaalwetenschappelijke problemen en zal in de komende versie van VASP wereldwijd beschikbaar komen voor onderzoekers.
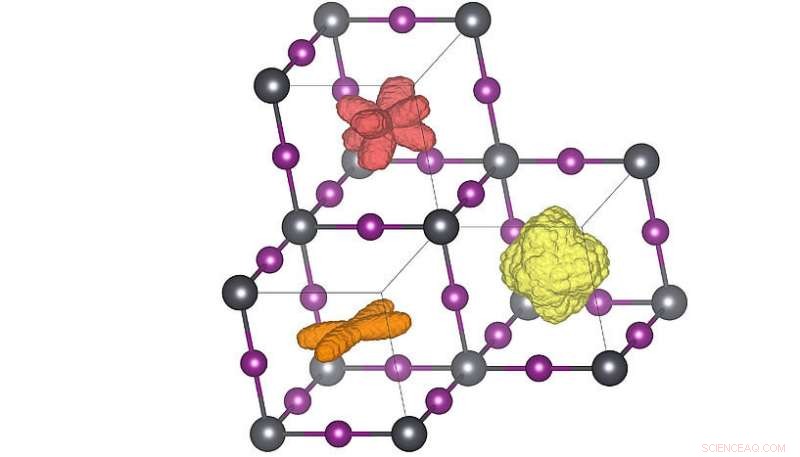
Driedimensionale verdelingen van de oriëntatie van het molecuul in de drie verschillende kristalfasen. Wanneer de temperatuur wordt verhoogd (oranje → rood → geel) kunnen de moleculen meer oriëntaties bereiken. De rode verdeling komt overeen met de structuur van de kamertemperatuur. Credit:Menno Bokdam/Universiteit van Wenen
 Hoe valentie van elektronen in het periodiek systeem te berekenen
Hoe valentie van elektronen in het periodiek systeem te berekenen - Onderzoek kijkt naar wrijvingseigenschappen van materiaal
- Onderzoekers ontwikkelen groene alternatieven voor fossiele grondstoffen
- Vroege ziektediagnose kan drastisch worden verbeterd met nieuw detectiesysteem
- Onderzoekers ontdekken chemische reactie die een verrassend molecuul gebruikt
Hoofdlijnen
- Onderzoeker bespreekt de biologische overspraak tussen microben en gastheren
- Fun Biology Presentatie Onderwerpen
- Biogeografie: definitie, theorie, bewijs & voorbeelden
- Ideeën voor het maken van een 3-D DNA-standaard voor middelbare school
- Het overdrachtspotentieel van vliegen kan groter zijn dan gedacht, onderzoekers zeggen:
- Wat zijn dromen?
- Vasculaire planten: definitie, classificatie, kenmerken en voorbeelden
- Is het kennen van je volledige genoom een recht of een voorrecht?
- Hoe een omgedraaid gen vlinders hielp mimicry te ontwikkelen
- Realtime 3D-reconstructie van complexe scènes van lange afstanden geven vorm aan ons heden en onze toekomst
- Eerste magneet geïnstalleerd voor het ALPS II-experiment bij DESY

- Uniforme theorie legt uit hoe materialen transformeren van vaste stoffen naar vloeistoffen

- Doe de twist:tweedimensionale kwantummaterialen maken met gebogen oppervlakken

- Door mist snijden met laserfocus


 3-fasig vermogen berekenen
3-fasig vermogen berekenen - Drones maken de eerste gedetailleerde kartering mogelijk van het Hoge Plateaus Basin in de Marokkaanse Atlas
- Tesla zonder Musk aan het stuur? Dat is wat de SEC wil
- Nieuw onderzoek onthult hoe sterrenstelsels warm en gehinderd blijven
- Verstikkende rook boven San Francisco zorgt voor vertragingen, sluitingen
- Zwitserse autoriteiten waarschuwen voor instorting gletsjer gebied evacueren
- Toegenomen smeltende sneeuw in de winter bedreigt westerse watervoorraden
- Een openbare energie-industrie kan helpen energiearmoede aan te pakken en hernieuwbare energiebronnen te vergroten
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
