Wetenschap
Evolutionair algoritme genereert op maat gemaakte moleculaire vingerafdrukken
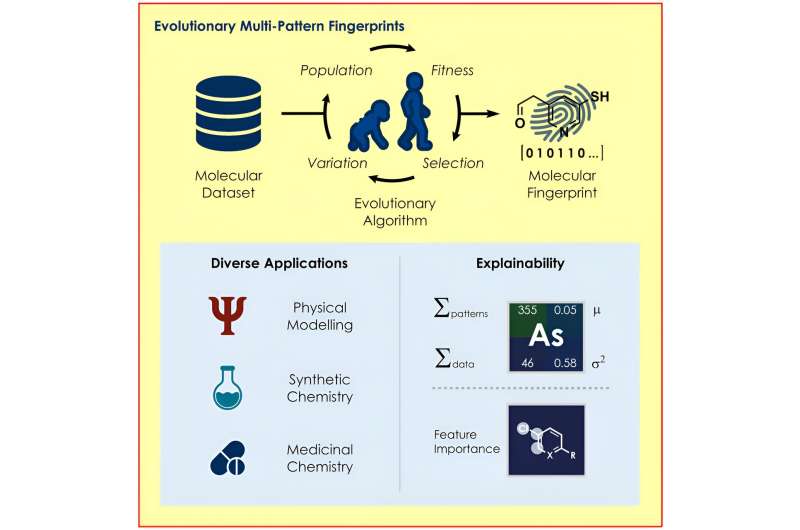
Een team onder leiding van prof. Frank Glorius van het Instituut voor Organische Chemie van de Universiteit van Münster heeft een evolutionair algoritme ontwikkeld dat de structuren in een molecuul identificeert die bijzonder relevant zijn voor een bepaald vraagstuk en deze gebruikt om de eigenschappen van de moleculen voor verschillende doeleinden te coderen. modellen voor machinaal leren.
De methode is ook geschikt voor het machinaal voorspellen van kwantumchemische eigenschappen en de toxiciteit van moleculen. Het kan worden toegepast op elke moleculaire dataset en vereist geen deskundige kennis van de onderliggende relaties.
Kunstmatige intelligentie en machinaal leren worden steeds relevanter in het dagelijks leven – en hetzelfde geldt voor de chemie. Organische chemici zijn bijvoorbeeld geïnteresseerd in hoe machinaal leren kan helpen bij het ontdekken en synthetiseren van nieuwe moleculen die effectief zijn tegen ziekten of op andere manieren nuttig zijn.
Het nieuwe algoritme, ontwikkeld door het team van Glorius, zoekt naar optimale moleculaire representaties op basis van de principes van de evolutie, met behulp van mechanismen zoals reproductie, mutatie en selectie. Afhankelijk van het model en de gestelde vraag worden op maat gemaakte ‘moleculaire vingerafdrukken’ gemaakt, die de scheikundigen in hun onderzoek gebruikten om chemische reacties met verrassende nauwkeurigheid te voorspellen.
De methode, gepubliceerd in het tijdschrift Chem , is ook geschikt voor het voorspellen van kwantumchemische eigenschappen en de toxiciteit van moleculen.
Om machinaal leren te kunnen gebruiken, moeten onderzoekers de moleculen eerst omzetten in een computerleesbare vorm. Veel onderzoeksgroepen hebben dit probleem al aangepakt en daarom zijn er verschillende manieren om deze taak uit te voeren. Het is echter moeilijk te voorspellen welke van de beschikbare methoden het meest geschikt is om een specifieke vraag te beantwoorden, bijvoorbeeld om te bepalen of een chemische verbinding schadelijk is voor de mens.
Het nieuwe algoritme is ontworpen om in elk geval de optimale moleculaire vingerafdruk te helpen vinden. Om dit te doen selecteert het algoritme geleidelijk de moleculaire vingerafdrukken die de beste resultaten behalen in de voorspelling uit vele willekeurig gegenereerde moleculaire vingerafdrukken.
"Naar het voorbeeld van de natuur gebruiken we mutaties, dat wil zeggen willekeurige veranderingen in individuele componenten van de vingerafdrukken, of combineren we componenten van twee vingerafdrukken", legt promovendus Felix Katzenburg uit.
"In andere onderzoeken worden moleculen vaak beschreven aan de hand van kwantificeerbare eigenschappen die door mensen zijn geselecteerd en berekend", voegt Glorius toe.
"Aangezien het algoritme dat we hebben ontwikkeld automatisch de relevante moleculaire structuren identificeert, zijn er geen systematische vooroordelen veroorzaakt door menselijke experts."
Een ander voordeel is dat de manier van coderen het mogelijk maakt om te begrijpen waarom een model een bepaalde voorspelling doet. Het is bijvoorbeeld mogelijk om conclusies te trekken over welke delen van een molecuul een positieve of negatieve invloed hebben op de voorspelling van hoe een reactie zal verlopen, waardoor onderzoekers de relevante structuren gericht kunnen veranderen.
Het team uit Münster ontdekte dat hun nieuwe methode niet altijd de meest optimale resultaten opleverde.
"Als er aanzienlijke menselijke expertise is gestoken in het selecteren van bijzonder relevante moleculaire eigenschappen of als er zeer grote hoeveelheden gegevens beschikbaar zijn, hebben andere methoden, zoals neurale netwerken, soms een voorsprong", zegt Katzenburg.
Een van de belangrijkste doelstellingen van het onderzoek was echter het ontwikkelen van een methode voor het coderen van moleculen die kan worden toegepast op elke moleculaire dataset en waarvoor geen deskundige kennis van de onderliggende relaties vereist is.