Wetenschap
Een nieuwe Hongaarse methode kan eiwitonderzoek helpen
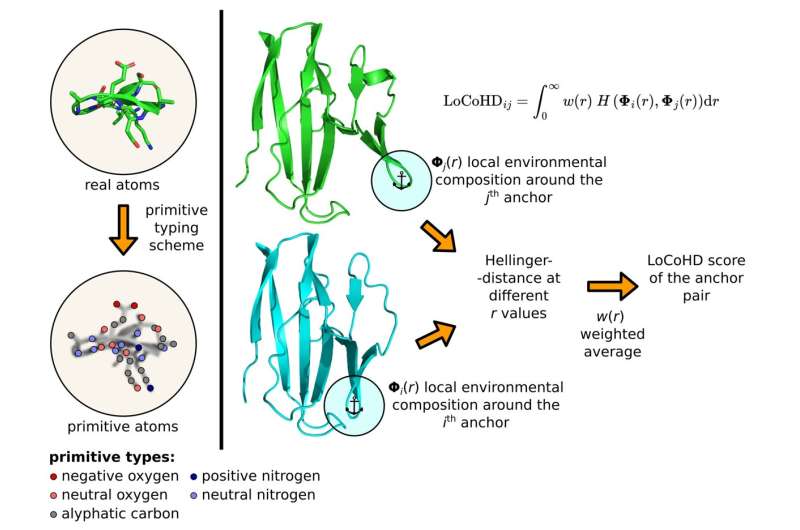
In een artikel dat onlangs is gepubliceerd in Nature Communications heeft de HUN-REN-ELTE Protein Modeling Research Group (Institute of Chemistry) de basis gelegd voor een wiskundige methode, die de computerondersteunde vergelijking van de driedimensionale structuren van eiwitten mogelijk maakt. De methode is uniek omdat, terwijl de tot nu toe beschikbare alternatieven alleen rekening hielden met de positie van de atomen, de nieuwe techniek, genaamd LoCoHD (Local Composition Hellinger Distance), ook de chemische informatie van de atomen omvat.