Wetenschap
Spinchemie heroverwegen vanuit een kwantumperspectief
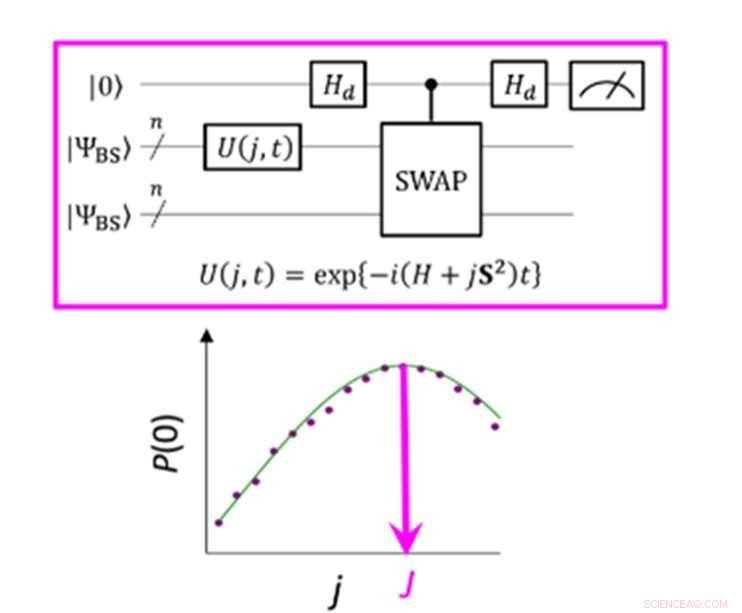
Een kwantumcircuit dat de maximale waarschijnlijkheid van P(0) mogelijk maakt bij het meten van de parameter J. Credit:K. Sugisaki, K. Sato en T. Takui
Onderzoekers van de Osaka City University gebruiken kwantumsuperpositietoestanden en Bayesiaanse gevolgtrekkingen om een kwantumalgoritme te creëren. gemakkelijk uitvoerbaar op kwantumcomputers, dat nauwkeurig en direct energieverschillen berekent tussen de elektronische grond en de aangeslagen spintoestanden van moleculaire systemen in polynomiale tijd.
Als we begrijpen hoe de natuurlijke wereld werkt, kunnen we deze nabootsen ten behoeve van de mensheid. Bedenk eens hoeveel we afhankelijk zijn van batterijen. De kern is het begrijpen van moleculaire structuren en het gedrag van elektronen daarin. Het berekenen van de energieverschillen tussen de elektronische grond van een molecuul en de aangeslagen spintoestanden helpt ons te begrijpen hoe we dat molecuul beter kunnen gebruiken in een verscheidenheid aan chemische, biomedische en industriële toepassingen. We hebben veel vooruitgang geboekt in moleculen met gesloten-schaalsystemen, waarin elektronen gepaard en stabiel zijn. Open-schaal systemen, anderzijds, zijn minder stabiel en hun onderliggende elektronische gedrag is complex, en daardoor moeilijker te begrijpen. Ze hebben ongepaarde elektronen in hun grondtoestand, die ervoor zorgen dat hun energie varieert vanwege de intrinsieke aard van elektronenspins, en maakt metingen moeilijk, vooral als de moleculen in omvang en complexiteit toenemen. Hoewel dergelijke moleculen in de natuur overvloedig aanwezig zijn, er is een gebrek aan algoritmen die deze complexiteit aankunnen. Een hindernis was het omgaan met wat de exponentiële explosie van rekentijd wordt genoemd. Het zou honderden miljoenen jaren duren om een conventionele computer te gebruiken om te berekenen hoe de ongepaarde spins de energie van een molecuul met een open schil beïnvloeden. tijd die mensen niet hebben.
Quantumcomputers zijn in ontwikkeling om dit te helpen reduceren tot wat 'polynomiale tijd' wordt genoemd. Echter, het proces dat wetenschappers hebben gebruikt om de energieverschillen van moleculen met een open schil te berekenen, is in wezen hetzelfde geweest voor zowel conventionele als kwantumcomputers. Dit belemmert het praktische gebruik van quantum computing in chemische en industriële toepassingen.
"Benaderingen die echte kwantumalgoritmen aanroepen, helpen ons open-shell-systemen veel efficiënter te behandelen dan door klassieke computers te gebruiken, ", stellen Kenji Sugisaki en Takeji Takui van de Osaka City University. Met hun collega's, ontwikkelden ze een kwantumalgoritme dat uitvoerbaar is op kwantumcomputers, welke kan, Voor de eerste keer, nauwkeurig energieverschillen berekenen tussen de elektronische grond en de aangeslagen spintoestanden van moleculaire systemen met open schil. Hun bevindingen werden gepubliceerd in het tijdschrift Chemische Wetenschappen op 24 december 2020.
Het energieverschil tussen moleculaire spintoestanden wordt gekenmerkt door de waarde van de uitwisselingsinteractieparameter J. Conventionele kwantumalgoritmen hebben nauwkeurig energieën kunnen berekenen voor moleculen met een gesloten schil "maar ze zijn niet in staat geweest om systemen met een sterke multi-configurationele karakter, " stelt de groep. Tot nu toe, wetenschappers hebben aangenomen dat om de parameter J te verkrijgen, eerst de totale energie van elke spintoestand moet worden berekend. Bij moleculen met een open schil is dit moeilijk omdat de totale energie van elke spintoestand sterk varieert naarmate het molecuul verandert in activiteit en grootte. Echter, "het energieverschil zelf is niet erg afhankelijk van de systeemgrootte, " merkt het onderzoeksteam op. Dit bracht hen ertoe een algoritme te maken met berekeningen die gericht waren op het spinverschil, niet de individuele spintoestanden. Het creëren van een dergelijk algoritme vereiste dat ze de aannames loslieten die waren ontwikkeld na jarenlang gebruik van conventionele computers en zich moesten concentreren op de unieke kenmerken van kwantumcomputing, namelijk 'quantumsuperpositietoestanden'.
"Superpositie" laat algoritmen twee variabelen tegelijk vertegenwoordigen, waardoor wetenschappers zich vervolgens kunnen concentreren op de relatie tussen deze variabelen zonder eerst hun individuele toestand te hoeven bepalen. Het onderzoeksteam gebruikte iets dat een golffunctie met gebroken symmetrie wordt genoemd als een superpositie van golffuncties met verschillende spintoestanden en herschreef het in de Hamiltoniaanse vergelijking voor de parameter J. Door dit nieuwe kwantumcircuit uit te voeren, het team was in staat om zich te concentreren op afwijkingen van hun doel en door Bayesiaanse gevolgtrekking toe te passen, een machine learning-techniek, ze brachten deze afwijkingen in om de uitwisselingsinteractieparameter J te bepalen. "Numerieke simulaties op basis van deze methode werden uitgevoerd voor de covalente dissociatie van moleculaire waterstof (H 2 ), de drievoudige bindingsdissociatie van moleculaire stikstof (N 2 ), en de grondtoestanden van C, O, Si-atomen en NH, OH + , CH 2 , NF en O 2 moleculen met een fout van minder dan 1 kcal/mol, ", voegt het onderzoeksteam toe.
"We zijn van plan onze Bayesiaanse eXchange-koppelingsparametercalculator met Broken-symmetry Wave Functions (BxB) -software te installeren op quantumcomputers voor de korte termijn die zijn uitgerust met ruis (geen kwantumfoutcorrectie) tussenschaal (enkele honderden qubits) kwantumapparaten (NISQ-apparaten) ), het testen van het nut voor kwantumchemische berekeningen van werkelijke omvangrijke moleculaire systemen."
 Als u azijn verdunt, hoe beïnvloedt dit de pH-waarde?
Als u azijn verdunt, hoe beïnvloedt dit de pH-waarde? - Welk eiwit is dat?
- De grenzen van wrijving voorspellen:team kijkt naar materiaaleigenschappen
- Superflexibele aerogels zijn zeer efficiënte absorptiemiddelen, thermische isolatoren, en druksensoren
- Langlevende katalysator vermindert dieselemissies
- Giftige algen nemen toe in Lake Okeechobee in Florida
- Plantendiversiteit slachtoffer van zeer ernstige bosbranden
- De impact van diepzeemijnbouw begrijpen
- NASA-stagiairs helpen het terminatorprobleem op te lossen via GLOBE challenge
- Aardbeving met een kracht van 6,7 op de schaal van Richter treft Filipijnen:USGS
Hoofdlijnen
- Hoe het Jeruzalem-syndroom werkt
- Niet alle kroontjeskruid is gelijk voor eierleggende monarchen, studie onthult
- Maïsplaag maakt gebruik van plantafweerstoffen om zichzelf te beschermen
- Nieuwe genen op verslechterend Y-chromosoom
- Bacteriën: definitie, soorten en voorbeelden
- Wat zijn de rollen van chlorofyl A & B?
- Lijst van forensische technieken
- Het magische medicijn van Bezoars
- Wat is de elektrische impuls die een Axon naar beneden beweegt?
- Neutronenmicelmetingen geven inzicht in verbeterde medicijnafgifte

- Atomair dunne perovskieten boost voor toekomstige elektronica

- Wetenschappers suggereren een nieuwe methode voor het synthetiseren van een veelbelovend magnetisch materiaal

- Wetenschappers verkrijgen met succes een synthetische groeifactor die compatibel is met het inheemse eiwit
- Wasbaar elektronisch textiel om een tijdperk van nog slimmere draagbare producten in te luiden


 Onderzoekers schetsen beleidsbenaderingen om brandbeheer te transformeren
Onderzoekers schetsen beleidsbenaderingen om brandbeheer te transformeren- Ammoniak op aanvraag? Alternatieve productiemethode voor een duurzame toekomst
- Wetenschappers ontdekken dat mechanisch gedrag van kleine structuren wordt beïnvloed door atoomdefecten
- Waarom is het voedselweb belangrijk?
- Hoe GPM te converteren naar HP
- Observeren en besturen van ultrasnelle processen met een resolutie van attoseconden
- Kleine schelpen duiden op grote veranderingen in de wereldwijde koolstofcyclus
- Inzicht in de omslagpunten van klimaatverandering uit het verleden kan ons helpen ons voor te bereiden op de toekomst
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
