Wetenschap
Actief leren versnelt het ontdekken van redox-flow-batterijen
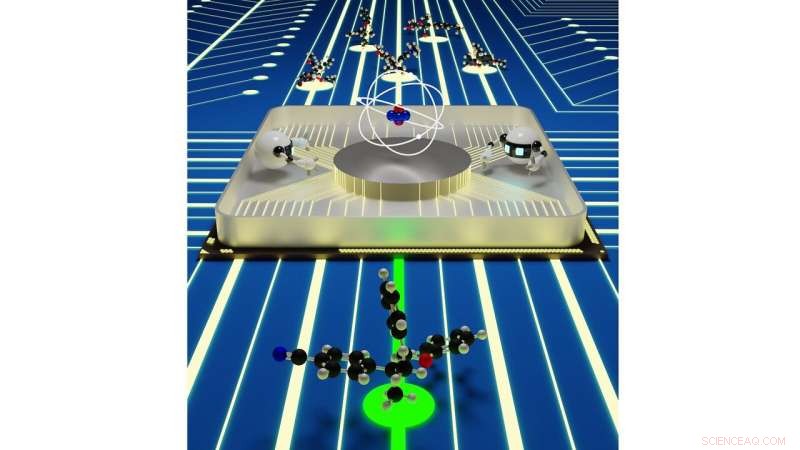
Naadloze interacties tussen kwantummechanische simulaties en kunstmatige intelligentie zouden een efficiënt platform voor het ontdekken van materialen kunnen bieden. Krediet:Rajeev Surendran Assary / Argonne National Laboratory
Door actief leren te gebruiken, wetenschappers vinden sneller geschikte kandidaten voor redox-flow-batterijen.
Als het tijd is om een nieuwe batterijchemie te ontwerpen, wetenschappers kunnen slechts een handvol mogelijkheden experimenteel uitproberen, omdat het tijd en middelen kost om elk nieuw molecuul te synthetiseren en te onderzoeken. Door betrouwbare moleculaire simulaties uit te voeren met behulp van supercomputers, onderzoekers kunnen het gewenste materiaalscreeningproces versnellen en hun zoektocht verbreden, terwijl u gedetailleerde informatie krijgt over de mogelijkheden die inherent zijn aan verschillende chemieën.
Echter, zelfs simulaties met hoge doorvoer die op deze supercomputers worden uitgevoerd, kunnen slechts een fractie van de mogelijke levensvatbare chemie voor bepaalde soorten batterijen bekijken. In een nieuwe studie van het Argonne National Laboratory van het Amerikaanse Department of Energy (DOE), onderzoekers zetten de volgende stap in het versnellen van de jacht op de best mogelijke batterijcomponenten door gebruik te maken van kunstmatige intelligentie.
Het studieteam, onder leiding van Argonne-chemicus Rajeev Surendran Assary, de innerlijke werking van redoxflow-batterijen onderzocht, waarin chemische energie wordt opgeslagen in opgeloste moleculen die interageren met elektroden. Flowbatterijen zijn kansrijk voor toepassingen in het elektriciteitsnet. Ze vervangen vaste kathoden en anodes door vloeibare oplossingen die doordrenkt zijn met moleculen die energie opslaan en vrijgeven. Conventionele stroombatterijen zijn gebaseerd op moleculen die één ladingsopslagelement per molecuul hebben, met beperkte veelzijdigheid. Onderzoekers van het Joint Centre for Energy Storage Research (JCESR), een DOE Energy Innovation Hub onder leiding van Argonne, introduceerde het concept van het opslaan en vrijgeven van energie met materialen genaamd "redox actieve polymeren, " of redoxmeren, die gebaseerd zijn op grotere moleculen, elk met tientallen ladingsopslagelementen.
In vergelijking met conventionele systemen, redoxmers bieden veel meer flexibiliteit om veel aspecten van batterijkenmerken en prestaties onafhankelijk aan te passen. Redoxmer-stroombatterijen openen een nieuwe deur naar het ontwerp van stroombatterijen omdat ze hoge functionaliteit kunnen bieden tegen lage kosten, met weinig schade aan het milieu. JCESR's redoxmer-stroombatterijen hebben het potentieel om de manier waarop we denken over en het gebruik van stroombatterijen voor het elektriciteitsnet te transformeren.
In het geval van de onderzochte redoxmeren, Assary en zijn collega's merkten dat, als de batterij oplaadt en ontlaadt, ze hebben de neiging om een inactieve film te vormen. Om dit fenomeen te voorkomen, het Argonne-team wilde een redoxmer ontwerpen die bij een bepaalde spanning elektrisch kon worden gesplitst, het vrijmaken om opnieuw in de elektrolytoplossing te komen.
"Je kunt het zien als het schoonmaken van een pan waarop je kookt, " zei Argonne postdoctoraal onderzoeker Hieu Doan, een andere auteur van de studie. "Om kleverige voedselresten gemakkelijker te verwijderen, u kunt hoge temperaturen gebruiken, en dat doen we met elektriciteit."
De onderzoekers wilden de splijtspanning net buiten het normale werkvenster van de batterij hebben, zodat het de prestaties niet zou belemmeren, maar zou ook niet veel extra energie vergen.
Om een redoxmeer te vinden dat zou splijten bij de juiste spanning, Assary en het team wendden zich tot Argonne's Bebop-supercomputer in het Laboratory Computing Resource Center. Eerst, de onderzoekers liepen een set van 1, 400 verschillende redoxmeren met behulp van dichtheidsfunctionaaltheorie (DFT) berekeningen, die zeer nauwkeurig maar rekenkundig duur zijn. Echter, deze 1, 400 redoxmers vertegenwoordigden slechts een klein deel van de totale chemische ruimte waarin de onderzoekers geïnteresseerd waren.
"Experimenteel, het kan maanden duren om een dozijn van deze redoxmeren te synthetiseren en te testen, dus het is essentieel om meer dan duizend redoxmers op de computer in detail te kunnen bestuderen, ' zei Assary.
Elk van deze redoxmeren bestaat uit een moleculaire steiger waarop een verscheidenheid aan verschillende chemische functionele groepen is geplaatst - die extra atomen of moleculen zijn. "De steiger is ontworpen op basis van suggesties van onze experimentele medewerkers, " zei Doan. Hoewel de steiger consistent is over de redoxmers, het variëren van de functionele groepen geeft verschillende eigenschappen.
Om de ideale moleculen te vinden uit een grotere dataset bestaande uit meer dan 100, 000 redoxmers zonder uitgebreide DFT-berekeningen, de onderzoekers gebruikten een machine learning-techniek genaamd actief leren. Deze grotere dataset bevatte redoxmeren die qua structuur vergelijkbaar waren met die in de oorspronkelijke DFT-dataset van 1. 400 moleculen - voor zover beide sets moleculen dezelfde steiger gebruikten. Echter, vanwege de verschillende manieren waarop de functionele groepen werden bevolkt, de eigenschappen liepen uiteen.
"Hoeveel leren u kunt doen in machine learning, wordt beperkt door uw trainingsdataset, "Zei Assary. "Je kunt alleen weten wat je hebt gezien, en als je iets anders hebt waar je voorspellingen over probeert te doen, het is misschien niet effectief."
In plaats van te trainen op het geheel van de gegevens, Assary en zijn collega's trainden het model op slechts een handvol verschillende redoxmer-mogelijkheden. Volgens Doan, na het trainen van het model met 10 datapunten, het model kiest zelf het 11e datapunt uit de resterende datapool.
"Het model garandeert dat door dit nieuwe datapunt toe te voegen aan de trainingsset, het zal beter worden, en dan kunnen we het weer trainen, "Zei Doan. "Wat de nauwkeurigheid van het model ook maximaliseert, dat zal het volgende datapunt zijn om te kiezen."
Assary zei dat om 30 moleculen met de gewenste eigenschappen te identificeren uit een initiële dataset van 1, 400, nam slechts 70 keuzes. Met willekeurige selectie, slechts 9 procent van de keuzes zou succesvol zijn geweest, een vijfvoudige verbetering betekent.
"Een aanzienlijke verbetering ten opzichte van zo'n grote chemische ruimte is opmerkelijk, "zei Assary. Inderdaad, toen dezelfde benadering werd toegepast op de 100, 000+ dataset, het vond met succes 42 gewenste moleculen binnen 100 picks.
Een paper gebaseerd op de studie, "Kwantumchemie-geïnformeerd actief leren om het ontwerp en de ontdekking van duurzame materialen voor energieopslag te versnellen, " werd gepubliceerd in het nummer van 28 mei van Chemie van materialen .
 Fatale horizon, gedreven door verzuring, sluit zich aan bij mariene organismen in de Zuidelijke Oceaan
Fatale horizon, gedreven door verzuring, sluit zich aan bij mariene organismen in de Zuidelijke Oceaan- Australische staat pompt zuurstof in rivieren als vissen sterven
- Nutriëntenrecyclerende microben kunnen de hitte voelen
- Jaar van extremen voor krimpende Zwitserse gletsjers in 2018:studie
- Nieuwe aanpak om gifstoffen uit afvalwater te verwijderen
Hoofdlijnen
- De ellende van Merkel neemt toe als minister van Landbouw orders negeert (update)
- Chemische stoffen gebruikt in de forensische wetenschap
- Passagiersduivengenoom toont effecten van natuurlijke selectie in een enorme populatie
- Stappen voor het traceren van een plant:zaadverspreiding volgen met behulp van chloroplast-DNA
- Verbazingwekkende diversiteit aan soorten gerapporteerd over Solent oesterrestauratieproject
- Welke bijdrage heeft Avery geleverd aan de ontdekking van DNA?
- Elements of Nucleic Acids
- Kleine wespenlichamen betekent kleine hersengebieden van wespen, studie toont
- De celstructuur van een ui
- Verhelderend hoe stikstofase ammoniak maakt
- Röntgenlaserwetenschappers ontwikkelen een nieuwe manier om te zien hoe bacteriën antibiotica aanvallen

- Ontdekken wat organellen met elkaar verbindt, kan helpen bij het begrijpen van neurodegeneratieve ziekten
- NREL onderzoekt coatings die nodig zijn voor het concentreren van zonne-energie

- Grote stappen voor het vastleggen van koolstof met behulp van aarde-overvloedige elementen als fotokatalytisch systeem

 Vissen profiteren als grote dammen een nieuwe manier van werken aannemen
Vissen profiteren als grote dammen een nieuwe manier van werken aannemen- Rapport:populaire VAE-chat-app ToTok een spionagetool van de overheid
- Bossen planten kan de planeet meer afkoelen dan gedacht
- Airbus zet 3,6 miljard euro opzij voor schikking corruptiezaak
- Magma-oceaan is mogelijk verantwoordelijk voor het vroege magnetische veld van de manen
- Welke drie manieren zijn dieren belangrijk voor planten?
- Het voorspellen van de rij-persoonlijkheden van mensen
- EPCOT was Walt Disneys radicale visie voor een nieuw soort stad
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
