Wetenschap
Machine learning begeleidt koolstofnanotechnologie
Koolstofnanostructuren zouden gemakkelijker te ontwerpen en te synthetiseren kunnen zijn dankzij een machine learning-methode die voorspelt hoe ze groeien op metalen oppervlakken. De nieuwe aanpak, ontwikkeld door onderzoekers van de Japanse Tohoku Universiteit en de Chinese Shanghai Jiao Tong Universiteit, zal het gemakkelijker maken om de unieke chemische veelzijdigheid van koolstofnanotechnologie te benutten. De methode is gepubliceerd in het tijdschrift Nature Communications .
De groei van koolstofnanostructuren op verschillende oppervlakken, waaronder als atomair dunne films, is uitgebreid bestudeerd, maar er is weinig bekend over de dynamiek en factoren op atomair niveau die de kwaliteit van de resulterende materialen bepalen. "Ons werk richt zich op een cruciale uitdaging voor het realiseren van het potentieel van koolstofnanostructuren in elektronica of energieverwerkingsapparatuur", zegt Hao Li van het team van Tohoku University.
Het brede scala aan mogelijke oppervlakken en de gevoeligheid van het proces voor verschillende variabelen maken direct experimenteel onderzoek een uitdaging. De onderzoekers wendden zich daarom tot machine learning-simulaties als een effectievere manier om deze systemen te verkennen.
Met machinaal leren kunnen verschillende theoretische modellen worden gecombineerd met gegevens uit scheikundige experimenten om de dynamiek van de kristallijne groei van koolstof te voorspellen en te bepalen hoe deze kan worden gecontroleerd om specifieke resultaten te bereiken. Het simulatieprogramma onderzoekt strategieën en identificeert welke werken en welke niet, zonder dat mensen elke stap van het proces moeten begeleiden.
-
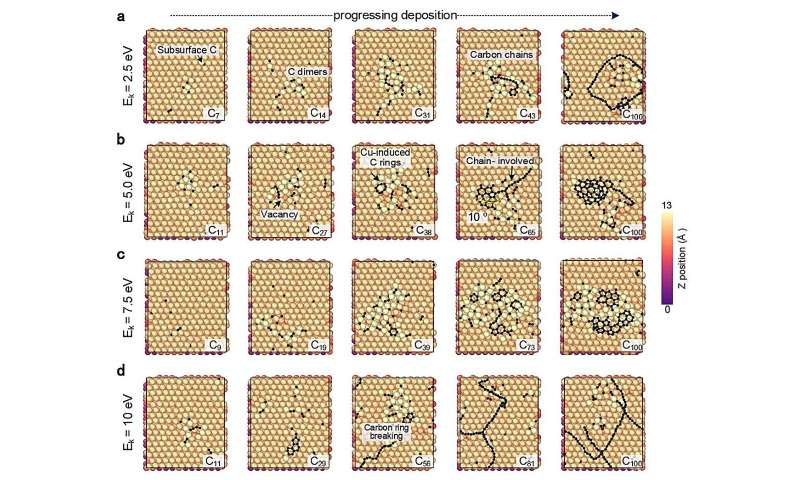
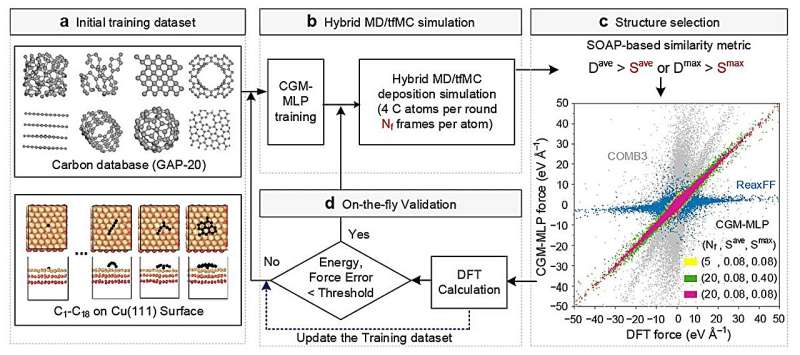
CGM-MLP-gestuurde simulaties van grafeengroei op Cu(111) met verschillende kinetische energieën voor koolstofincidenten (Ek). (a) 2,5 eV, (b) 5,0 eV, (c) 7,5 eV en (d) 10 eV. Credit:Natuurcommunicatie (2024). DOI:10.1038/s41467-023-44525-z -
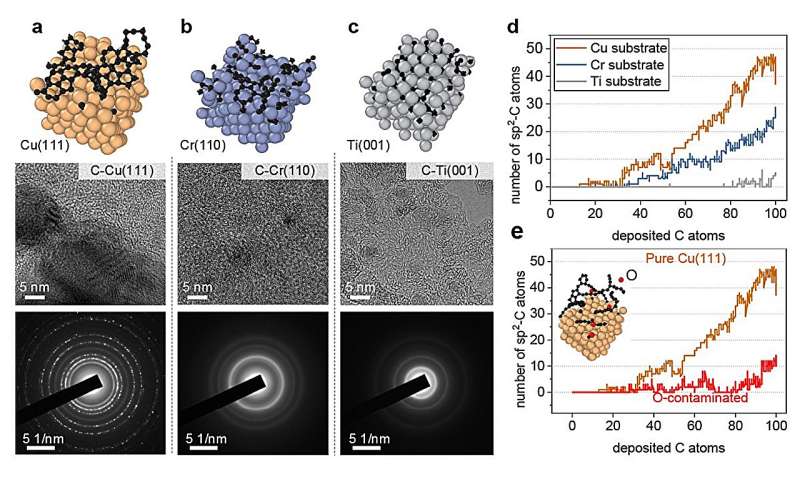
Representatieve metalen oppervlakken voor de groei van koolstofnanostructuren. (a) zuiver Cu(111), (b) Cr(110 en (c) Ti(001) oppervlak. Onder elk oppervlak, hoge resolutie transmissie-elektronenmicroscopie (HRTEM) beelden en geselecteerde gebied elektronendiffractie (SAED) beelden van koolstof nanostructuren die zijn vervaardigd door depositie door magnetronsputteren worden gegeven. (d) Het aantal sp 2 -C als functie van afgezette koolstofatomen op verschillende metaalsubstraten en e O-verontreinigd Cu(111). Credit:Natuurcommunicatie (2024). DOI:10.1038/s41467-023-44525-z
De onderzoekers testten deze aanpak door simulaties te onderzoeken van de groei van grafeen, een vorm van koolstof, op een koperoppervlak. Nadat ze het basiskader hadden vastgesteld, lieten ze zien hoe hun aanpak ook kon worden toegepast op andere metalen oppervlakken, zoals titanium, chroom en koper die verontreinigd zijn met zuurstof.
De verdeling van elektronen rond de kernen van atomen in verschillende vormen van grafeenkristallen kan variëren. Deze subtiele verschillen in atomaire structuur en elektronenrangschikking beïnvloeden de algehele chemische en elektrochemische eigenschappen van het materiaal. De machine learning-aanpak kan testen hoe deze verschillen de diffusie van individuele atomen en gebonden atomen en de vorming van koolstofketens, bogen en ringstructuren beïnvloeden.
Het team valideerde de resultaten van de simulaties door middel van experimenten en ontdekte dat ze nauw bij elkaar pasten. "Over het geheel genomen biedt ons werk een praktische en efficiënte methode voor het ontwerpen van metalen of gelegeerde substraten om de gewenste koolstofnanostructuren te bereiken en verdere mogelijkheden te verkennen", zegt Li.
Hij voegt eraan toe dat toekomstig werk hierop zal voortbouwen om onderwerpen te onderzoeken zoals de grensvlakken tussen vaste stoffen en vloeistoffen in geavanceerde katalysatoren en de chemische eigenschappen van materialen die worden gebruikt voor de verwerking en opslag van energie.
Meer informatie: Di Zhang et al, Actief machinaal leermodel voor de dynamische simulatie- en groeimechanismen van koolstof op metaaloppervlak, Nature Communications (2024). DOI:10,1038/s41467-023-44525-z
Aangeboden door Tohoku Universiteit
 Concurrerende krachten:hoe moleculen hun structuur behouden?
Concurrerende krachten:hoe moleculen hun structuur behouden?- Een magnetische kurk voor het verwijderen van watervervuiling
- Hoe bewaar je een ei in azijn voor een wetenschapsproject om een ei in een fles te krijgen
- Een goedkope vervanger voor duur metaal in een industrieel gebruikelijke chemische reactie
- Onderzoekers meten elektronenemissie om het begrip van lasergebaseerd 3D-printen van metaal te verbeteren
Hoofdlijnen
- Haaienvinnenverboden helpen haaien misschien niet, wetenschappers zeggen:
- Waarom walvissen geen hersenbeschadiging oplopen als ze zwemmen
- Vliegen ontwikkelen springen zonder poten
- Landbouwgroepen dagen waarschuwing onkruidverdelger in Californië uit
- Wat is de schijfachtige structuur aan de zijde van chloroplasten?
- Antibioticavervuiling verstoort het darmmicrobioom en blokkeert het geheugen van waterslakken, zo blijkt uit onderzoek
- De controversiële handel in bushmeat herkaderen:wie bepaalt welke voedingsmiddelen geschikt zijn voor consumptie?
- Fun Biology Presentatie Onderwerpen
- De nieuwste meren van Alaska boeren methaan op
- Wetenschappers maken apparaat dat een licht pincet gebruikt om virussen te vangen en te verplaatsen

- Voedingsvezels zuiveren effectief koolstofnanobuisjes

- Gouden nanodeeltjes om in-situ detectie van geamplificeerd DNA bij kamertemperatuur te vergemakkelijken

- De ontvlambaarheid van epoxyhars in bedwang houden

- Interactie van koolstofnanobuisjes en de bloed-hersenbarrière

 Soorten Silverfish
Soorten Silverfish- Voor het eerst in 5 jaar, Amerikaanse benzineverbruik gedaald, uitstoot omhoog
- Berekening van pompgalons per minuut
- CHIME-telescoop detecteert meer dan 500 mysterieuze snelle radio-uitbarstingen in het eerste jaar van gebruik
- Het opruimen van verlaten mijnen betekent dat we allemaal de prijs betalen
- Rapport identificeert grote uitdagingen om beter voor te bereiden op vulkaanuitbarstingen
- Grote mensenmassa's wachten op een totale zonsverduistering in Noord-Amerika. Wolken kunnen het uitzicht bederven
- Misschien vindt u een zeldzame soort in uw achtertuin:hoe mondiale burgerwetenschap bijdraagt aan kennis over biodiversiteit
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
