Wetenschap
Simulaties bieden een schat aan gefluoreerde verbindingen
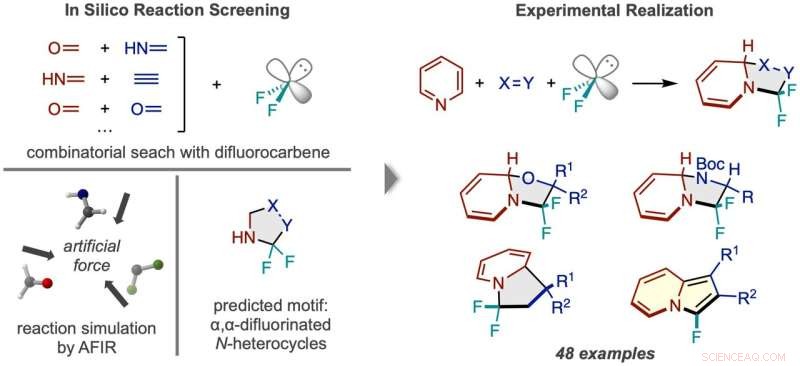
Workflow van reactiedetectie via in silico screening. (Links) Reacties tussen difluorcarbeen en talrijke paren kleine moleculen werden gesimuleerd, wat een N-heterocyclisch product voorspelde dat tweemaal gefluoreerd was aan de alfa-koolstof. (Rechts) Het succesvolle reactiekader met behulp van pyridine en voorbeelden van de soorten verkregen productverbindingen. Krediet:Natuursynthese (2022). DOI:10.1038/s44160-022-00128-y
Computersimulaties worden meestal als richtlijn gebruikt, zodat scheikundigen efficiënter de exacte details kunnen uitwerken van een algemeen reactie-idee dat ze in gedachten hebben - net zoals een kompas een ontdekkingsreiziger helpt efficiënt naar een bestemming op hun kaart te leiden. Onderzoekers van ICReDD gingen echter een grote stap verder en gebruikten simulaties om het algemene idee voor een volledig onvoorstelbare reactie te produceren, waarbij ze effectief berekeningen gebruikten om de kaart zelf te maken. Met behulp van het ontwerpprincipe dat wordt gesuggereerd door computationele resultaten, raakte het team de moederlode in het laboratorium en ontwikkelde met succes een reeks van 48 reacties die verbindingen produceren die mogelijk nuttig zijn voor de ontwikkeling van nieuwe geneesmiddelen.
De aanwezigheid en positie van fluor in een molecuul beïnvloedt vaak de farmacologische activiteit van een molecuul. Onderzoekers van ICReDD hebben kwantumchemische berekeningen gebruikt om een reactie te ontdekken die selectief twee fluoratomen toevoegt aan een moeilijk toegankelijke positie op een N-heterocyclus-moleculen met een koolstofringstructuur waarbij ten minste één koolstof in de ring is vervangen door stikstof . Het vermogen om fluoratomen te hechten aan de voorheen moeilijk toegankelijke "alfakoolstof" - de koolstof direct naast de stikstof in de ringstructuur - zou kunnen leiden tot de ontwikkeling van een groot aantal nieuwe medicijnen.
Voordat ze experimenten in het laboratorium uitvoerden, wierpen de onderzoekers een breed net uit, waarbij ze de levensvatbaarheid van talrijke 3-componentenreacties met behulp van de kunstmatige krachtgeïnduceerde reactie (AFIR) -methode computationeel testten. Ze simuleerden de reactie van een difluorcarbeenmolecuul, dat werkt bij de bron van fluoratomen, met verschillende paren kleine moleculen met een dubbele of driedubbele binding. Deze simulaties toonden aan dat een aantal ringvormende reacties levensvatbaar zouden moeten zijn.
Onderzoekers probeerden een van de veelbelovende reacties die werden gesuggereerd door de eerste computationele resultaten, maar waren niet succesvol. A more narrowly focused, optimized computation of the transition state energy of the reaction in question showed that the difluorocarbene molecule more easily reacted with itself than with the desired starting molecules, signaling that an undesired side reaction was likely occurring. This result inspired researchers to change one of the starting materials to the cyclic molecule pyridine, which they anticipated would be able to compete with the unwanted side reaction. This change resulted in the successful synthesis of the desired N-heterocyclic product with two fluorines attached at the alpha carbon position.
The reaction developed here is also significant because it breaks the aromatic system of electrons in the pyridine molecule, a transformation that is especially difficult due to the high stability of aromatic systems. Additionally, the 3-component reaction framework was applied successfully in the lab to a wide range of starting materials, resulting in many new molecules with unique alpha position fluorine substitutions. The large scope of reactivity greatly increases the potential utility of this reaction framework in new drug development.
The researchers see their streamlined screening method as a way to broaden the scope of their search and discover new horizons in chemical reaction design.
"Our study's highlight is the successful demonstration of an in silico reaction screening strategy for reaction development. The computational reaction simulation suggested less-explored three-component reactions of difluorocarbene and two unsaturated molecules, which we successfully realized in experiments," explained lead author Hiroki Hayashi.
"I think the AFIR method is a powerful tool for dictating new research directions in reaction discovery, and we plan to continue building a computation-based reaction development platform by integrating the computational and informatics techniques of ICReDD."
The study was published in Nature Synthesis . + Verder verkennen
Terugspoelen indrukken om chemische reacties in meerdere stappen te voorspellen
 Hoe snelheid te berekenen van temperatuur
Hoe snelheid te berekenen van temperatuur- Duurzame ijzerkatalyse maakt controleerbare alkeenborylering mogelijk
- Onderzoekers creëren eerste draagbare technologie voor het detecteren van cyanotoxinen in water
- Onderzoekers werpen nieuw licht op mysteries achter de lichtemissie van vuurvliegjes
- Ontkoppeling van elektronisch en thermisch transport
- Aan de kust van Tarragona, 57% van het plastic afval bestaat uit kledingvezels van wasmachines
- Fujitsu gebruikt 's werelds snelste supercomputer en AI om tsunami-overstromingen te voorspellen
- Onderzoekers gebruiken satellietbeelden om het grondwatergebruik in de centrale vallei van Californië in kaart te brengen
- Hoe meer overstromingen als gevolg van klimaatverandering de waterwegen in de VS beïnvloeden
- Misvattingen in de diepzee veroorzaken onderschatting van de effecten van mijnbouw op de zeebodem
Hoofdlijnen
- Wetenschappers ontwerpen bacteriën om sonarsignalen te reflecteren voor ultrasone beeldvorming
- Infectieziekten:CTRL + ALT + Delete
- Aan de Caribische kust van Mexico groeien bergen zeewier
- Wat zijn de waarschijnlijke resultaten van de nieuwe kennis van de mensheid over het menselijk genoom?
- Woordproblemen schrijven voor Math
- Hoe doodt alcohol bacteriën?
- Wasbloemen en hun complexe relatie
- Krachten van spinnengif onderzocht in VR-game
- Wat zijn de stadia van cytokinese?
- Zink reguleert de opslag en afgifte van neurotransmitters

- Onderzoekers identificeren de oorsprong van het metabolisme
- Spannende nieuwe ontwikkelingen voor polymeren gemaakt van afvalzwavel

- Een superieure, goedkope katalysator voor watersplitsing
- Nieuwe doorbraak opent deuren voor de behandeling van aan melanine gerelateerde huidaandoeningen

 Een nieuwe methode om waterstof efficiënter uit water te halen om duurzame energie op te vangen
Een nieuwe methode om waterstof efficiënter uit water te halen om duurzame energie op te vangen- Is er een geheime stad onder Walt Disney World?
- Nieuwe Marsrover is klaar voor ruimtelasers
- Onderdelen van een drievoudige balkbalans & zijn toepassingen
- Hydrogen
- Nieuwe oplosmiddelen om plantaardige cellulose af te breken voor bio-ethanol
- Wapens Uitgevonden door Archimedes
- Wat zijn waterkrachtcentrales gemaakt van?
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
