Wetenschap
Nieuw Bayesiaans kwantumalgoritme berekent direct het energieverschil van een atoom en een molecuul
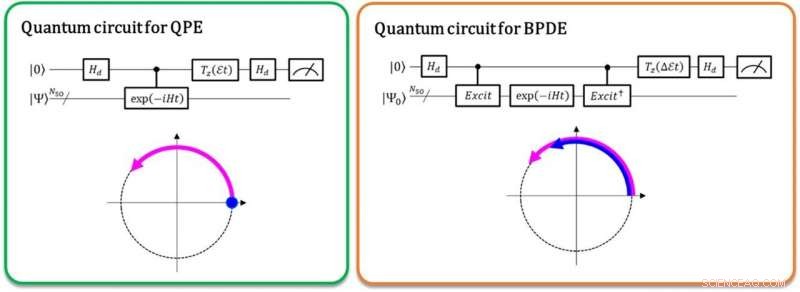
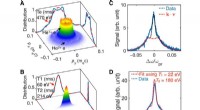
Links:Het faseverschil tussen |0⟩|Ψ⟩ en exp(-iEt)|1⟩|Ψ⟩ levert de totale energie E op. De gebogen pijl in paars geeft de fase-evolutie van |Ψ⟩ in de tijd aan. Rechts:Het faseverschil tussen exp(-iE0t)|0⟩|Ψ0 ⟩ en exp(-iE1t)|1⟩|Ψ1 ⟩ levert het energieverschil E1 - E0 op, direct. De gebogen pijlen in blauw en paars geven de fase-evolutie van |Ψ0 ⟩ en die van |Ψ1 ⟩ aan, respectievelijk. Krediet:K. Sugisaki, K. Sato en T. Takui
Zoals onlangs gemeld door het tijdschrift Fysische chemie Chemische fysica , onderzoekers van de Graduate School of Science aan de Osaka City University hebben een kwantumalgoritme ontwikkeld dat de elektronische toestanden van atomaire of moleculaire systemen kan begrijpen door het energieverschil in hun relevante toestanden direct te berekenen. Geïmplementeerd als een Bayesiaanse fase verschillende schatting, het algoritme breekt met de conventie door niet te focussen op het verschil in totale energieën berekend op basis van de pre- en postfase-evolutie, maar door de evolutie van het energieverschil zelf te volgen.
"Bijna alle scheikundeproblemen gaan over het energieverschil, niet de totale energie van het molecuul zelf, " zegt onderzoeksleider en speciaal aangestelde docent Kenji Sugisaki, "ook, moleculen met zware atomen die onderaan het periodiek systeem verschijnen, hebben grote totale energieën, maar de grootte van het energieverschil besproken in de chemie, zoals elektronische excitatietoestanden en ionisatie-energieën, hangt niet veel af van de grootte van het molecuul." Dit idee bracht Sugisaki en zijn team ertoe een kwantumalgoritme te implementeren dat direct energieverschillen berekent in plaats van totale energieën, het creëren van een toekomst waarin schaalbare of praktische kwantumcomputers ons in staat stellen om daadwerkelijk chemisch onderzoek en materiaalontwikkeling uit te voeren.
Momenteel, kwantumcomputers zijn in staat om de volledige configuratie-interactie (full-CI) berekeningen uit te voeren die optimale moleculaire energieën opleveren met een kwantumalgoritme genaamd kwantumfaseschatting (QPE), opmerkend dat de volledige CI-berekening voor omvangrijke moleculaire systemen onhandelbaar is met elke supercomputer. QPE vertrouwt op het feit dat een golffunctie, |Ψ⟩ die de wiskundige beschrijving van de kwantumtoestand van een microscopisch systeem aangeeft - in dit geval de wiskundige oplossing van de Schrödinger-vergelijking voor het microscopische systeem zoals een atoom of molecuul - verandert tijd-evolutief van fase afhankelijk van zijn totale energie. In de conventionele QPE, de kwantumsuperpositietoestand (|0⟩|Ψ⟩+|1⟩|Ψ⟩) ⁄ √2 wordt voorbereid, en de introductie van een operator voor gecontroleerde tijdevolutie zorgt ervoor dat |Ψ⟩ alleen in de tijd evolueert wanneer de eerste qubit de staat |1⟩ aangeeft. Dus, de |1⟩-toestand creëert een kwantumfase van de post-evolutie in de tijd, terwijl de |0⟩-toestand die van de pre-evolutie is. Het faseverschil tussen de pre- en post-evoluties geeft de totale energie van het systeem.
De onderzoekers van de Osaka City University generaliseren de conventionele QPE naar de directe berekening van het verschil in de totale energie tussen twee relevante kwantumtoestanden. In het nieuw geïmplementeerde kwantumalgoritme genaamd Bayesiaanse faseverschilschatting (BPDE), de superpositie van de twee golffuncties, (|0⟩|Ψ 0 ⟩ + |1⟩|Ψ 1 ) ⁄ √2, waar |Ψ 0 ⟩ en |Ψ 1 ⟩ geef de golffunctie aan die relevant is voor elke toestand, respectievelijk, is voorbereid, en het verschil in de fase tussen |Ψ 0 ⟩ en |Ψ 1 ⟩ na de tijd geeft de evolutie van de superpositie direct het verschil in de totale energie tussen de twee betrokken golffuncties. "We benadrukken dat het algoritme de evolutie van het energieverschil in de tijd volgt, die minder gevoelig is voor ruis dan het individueel berekenen van de totale energie van een atoom of molecuul. Dus, het algoritme voldoet aan de behoefte aan scheikundige problemen die nauwkeurige nauwkeurigheid in energie vereisen", stelt onderzoeksleider en emeritus professor Takeji Takui.
Eerder, deze onderzoeksgroep ontwikkelde een kwantumalgoritme dat direct het energieverschil berekent tussen elektronische toestanden (spintoestanden) met verschillende spinkwantumgetallen (K. Sugisaki, K.Toyota, K. Sato, D. Shiomi, T. Takui, Chem. Wetenschap. 2021, 12 , 2121-2132.). Dit algoritme, echter, vereist meer qubits dan de conventionele QPE en kan niet worden toegepast op de berekening van het energieverschil tussen de elektronische toestanden met gelijke spinkwantumgetallen, wat belangrijk is voor de spectrale toewijzing van UV-zichtbare absorptiespectra. Het in het onderzoek ontwikkelde BPDE-algoritme lost deze problemen op, waardoor het een zeer veelzijdig kwantumalgoritme is.
 Onderzoekers ontwikkelen hoogwaardige perovskietoxide-katalysatoren met behulp van laat-overgangsmetaaloxidematerialen
Onderzoekers ontwikkelen hoogwaardige perovskietoxide-katalysatoren met behulp van laat-overgangsmetaaloxidematerialen- Eigenschappen en gebruik van koolstofstaal
- Quantum wapening:Quantum dots verbeteren de stabiliteit van perovskietkristallen die zonne-energie oogsten
- Vloeibare kernvezels:er loopt een datarivier doorheen
- Polymeerkathode met hoge energiedichtheid voor snelladende natrium- en multivalent-ionbatterijen
Hoofdlijnen
- Onderzoeker bespreekt de biologische overspraak tussen microben en gastheren
- Onderzoekers ontsluiten mogelijk pad om vleesetende bacteriën te behandelen
- Middelgrote carnivoren lopen het grootste risico door veranderingen in het milieu
- Dominante fysische genen in Humans
- Kan een hersenscan je vertellen of je een crimineel gaat worden?
- Zijn er verschillen tussen mannelijke en vrouwelijke pesters?
- Epigenetische reostaat onthult hoe genregulatie wordt geërfd en onderhouden
- Hoe is het schrijven geëvolueerd?
- Trucs voor het onthouden van dieren Phylum
- Waarom 2D? Dikteafhankelijke elektronische eigenschappen meten
- Onstabiele rotspilaren in de buurt van reservoirs kunnen gevaarlijke watergolven veroorzaken

- Snellere LED's voor draadloze communicatie van onzichtbaar licht
- Ons begrip van di-fotonen verbeteren
- Ultrasnelle optisch veld-geïoniseerde gassen:een laboratoriumplatform om kinetische plasma-instabiliteiten te bestuderen

 Isopropanol Alcohol Vs. Isopropyl alcohol
Isopropanol Alcohol Vs. Isopropyl alcohol - Wat zit er onder de Antarctische ijskap?
- Onderzoekers identificeren recordaantal oude gereedschappen voor olifantenbot
- Ontdekking van het oudste zoogdier in Brazilië is een eerbetoon aan David Bowie
- De kosten van berekening
- voorbij Mars, het mini MarCO-ruimtevaartuig valt stil
- Edmunds kiest SUV's die veel brandstof verbruiken
- Wat is het Baader-Meinhof-fenomeen?
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
