Wetenschap
Machinaal leren doorbreekt het raadsel van de kwantumchemie

De tetraëdrische elektronische distributie van een watermolecuul. De zuurstofatoomkern bevindt zich in het midden van de tetraëder, en de waterstofkernen bevinden zich in het midden van de roze bollen. Stichting Simons. Krediet:Simons Foundation
Een nieuwe tool voor machine learning kan de energie berekenen die nodig is om een molecuul te maken of te breken, met een hogere nauwkeurigheid dan conventionele methoden. Hoewel de tool momenteel alleen eenvoudige moleculen aankan, het baant de weg voor toekomstige inzichten in de kwantumchemie.
"Het gebruik van machinaal leren om de fundamentele vergelijkingen op te lossen die de kwantumchemie beheersen, is al enkele jaren een open probleem, en er is nu veel opwinding omheen, " zegt mede-bedenker Giuseppe Carleo, een onderzoekswetenschapper aan het Centre for Computational Quantum Physics van het Flatiron Institute in New York City. Een beter begrip van de vorming en vernietiging van moleculen, hij zegt, zou de innerlijke werking van de chemische reacties die van vitaal belang zijn voor het leven kunnen onthullen.
Carleo en medewerkers Kenny Choo van de Universiteit van Zürich en Antonio Mezzacapo van het IBM Thomas J. Watson Research Center in Yorktown Heights, New York, presenteren hun werk 12 mei in Natuurcommunicatie .
De tool van het team schat de hoeveelheid energie die nodig is om een molecuul te assembleren of uit elkaar te trekken, zoals water of ammoniak. Die berekening vereist het bepalen van de elektronische structuur van het molecuul, die bestaat uit het collectieve gedrag van de elektronen die het molecuul aan elkaar binden.
De elektronische structuur van een molecuul is lastig te berekenen, waarbij de bepaling van alle potentiële toestanden van de elektronen van het molecuul nodig is, plus de kans van elke staat.
Omdat elektronen op elkaar inwerken en kwantummechanisch met elkaar verstrengeld raken, wetenschappers kunnen ze niet individueel behandelen. Met meer elektronen, meer verwikkelingen duiken op, en het probleem wordt exponentieel moeilijker. Exacte oplossingen bestaan niet voor moleculen die complexer zijn dan de twee elektronen in een paar waterstofatomen. Zelfs benaderingen worstelen met nauwkeurigheid wanneer ze meer dan een paar elektronen omvatten.
Een van de uitdagingen is dat de elektronische structuur van een molecuul toestanden bevat voor een oneindig aantal orbitalen die steeds verder van de atomen verwijderd zijn. Aanvullend, het ene elektron is niet van het andere te onderscheiden, en twee elektronen kunnen niet dezelfde toestand innemen. De laatste regel is een gevolg van uitwisselingssymmetrie, die bepaalt wat er gebeurt als identieke deeltjes van toestand veranderen.
Mezzacapo en collega's van IBM Quantum ontwikkelden een methode om het aantal overwogen orbitalen te beperken en uitwisselingssymmetrie op te leggen. Deze aanpak, gebaseerd op methoden die zijn ontwikkeld voor kwantumcomputertoepassingen, maakt het probleem meer verwant aan scenario's waarin elektronen zijn beperkt tot vooraf ingestelde locaties, zoals in een stijf rooster.
De gelijkenis met stijve roosters was de sleutel om het probleem beter beheersbaar te maken. Carleo heeft eerder neurale netwerken getraind om het gedrag van elektronen te reconstrueren die beperkt zijn tot de plaatsen van een rooster. Door deze methoden uit te breiden, de onderzoekers konden oplossingen inschatten voor de gecomprimeerde problemen van Mezzacapo. Het neurale netwerk van het team berekent de waarschijnlijkheid van elke toestand. Met behulp van deze kans, de onderzoekers kunnen de energie van een bepaalde toestand schatten. Het laagste energieniveau, de evenwichtsenergie genoemd, is waar het molecuul het meest stabiel is.
De innovaties van het team maakten het berekenen van de elektronische structuur van een basismolecuul eenvoudiger en sneller. De onderzoekers toonden de nauwkeurigheid van hun methoden aan door te schatten hoeveel energie het zou kosten om een molecuul uit de echte wereld uit elkaar te trekken, zijn banden verbreken. Ze voerden berekeningen uit voor diwaterstof (H 2 ), lithiumhydride (LiH), ammoniak (NH 3 ), water (H 2 O), diatomische koolstof (C 2 ) en distikstof (N 2 ). Voor alle moleculen, de schattingen van het team bleken zeer nauwkeurig, zelfs in gebieden waar bestaande methoden moeilijk zijn.
In de toekomst, de onderzoekers willen grotere en complexere moleculen aanpakken door meer geavanceerde neurale netwerken te gebruiken. Een van de doelen is het omgaan met chemicaliën zoals die in de stikstofcyclus worden aangetroffen, waarin biologische processen op stikstof gebaseerde moleculen bouwen en breken om ze bruikbaar te maken voor het leven. "We willen dat dit een hulpmiddel is dat door chemici kan worden gebruikt om deze problemen te verwerken, ' zegt Carleo.
Carleo, Choo en Mezzacapo zijn niet de enigen die machine learning gebruiken om problemen in de kwantumchemie aan te pakken. De onderzoekers presenteerden hun werk voor het eerst op arXiv.org in september 2019. In diezelfde maand een groep in Duitsland en een andere bij Google's DeepMind in Londen hebben elk onderzoek gepubliceerd met behulp van machine learning om de elektronische structuur van moleculen te reconstrueren.
De andere twee groepen gebruiken een vergelijkbare benadering van elkaar die het aantal overwogen orbitalen niet beperkt. Deze inclusiviteit, echter, rekent meer rekenkracht, een nadeel dat alleen maar erger wordt met complexere moleculen. Met dezelfde rekenkracht, de aanpak van Carleo, Choo en Mezzacapo levert een hogere nauwkeurigheid op, maar de vereenvoudigingen die zijn gemaakt om deze nauwkeurigheid te verkrijgen, kunnen vooroordelen introduceren.
"Algemeen, het is een afweging tussen vooringenomenheid en nauwkeurigheid, en het is onduidelijk welke van de twee benaderingen meer potentieel heeft voor de toekomst, " zegt Carleo. "Alleen de tijd zal ons leren welke van deze benaderingen kunnen worden opgeschaald naar de uitdagende open problemen in de chemie."
 Veelzijdige coating voor magnesium kan uiteindelijk leiden tot betere botimplantaten
Veelzijdige coating voor magnesium kan uiteindelijk leiden tot betere botimplantaten- Een moleculair afvalverwerkingscomplex speelt een rol bij het inpakken van het genoom
- Geneesmiddelen en producten voor persoonlijke verzorging opzuigen uit water
- Lang gezochte structuur van telomerase effent de weg voor nieuwe geneesmiddelen voor veroudering, kanker
- Duurzame optische vezels ontwikkeld uit methylcellulose
- Het Euraziatische continent onthoudt en versterkt koude golven naarmate het noordpoolgebied opwarmt
- Kenmerken en gedrag van de reuzenpanda
- Woestijnvorming en klimaatverandering door moesson gekoppeld aan verschuivingen in ijsvolume en zeeniveau
- Onderzoek ontsluit geheimen van ijzeropslag in algen
- Afschaffing van subsidies voor fossiele brandstoffen weinig hulp aan klimaat:studie
Hoofdlijnen
- Detectorhonden bieden hoop om numbats te redden
- Hoe beïnvloedt CO2 de opening van de huidmondjes?
- Het verschil tussen genomisch DNA en plasmide-DNA
- Het verschil tussen prokaryote en eukaryotische genexpressie
- Onderzoek onthult hoe verontreinigende stoffen de vroege embryonale ontwikkeling beïnvloeden
- Hoop voor honden met de meest voorkomende hart- en vaatziekten
- Onderzoeker ontdekt dat wanneer sperma concurreert, eieren hebben een keuze
- Ademhaling bij zoogdieren
- Afnemende babyzangvogels hebben bossen nodig om droogte te overleven
- "Bobcat 743 Specifications

- Onderzoekers vinden eerste overtuigend bewijs van nieuwe eigenschap die bekend staat als ferroelasticiteit in perovskieten
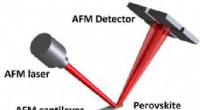
- Efficiënte generatie van relativistische mid-infraroodpulsen in de buurt van een cyclus in plasma's

- Veelzijdig LED-bestralingssysteem:van desinfectie tot medische behandelingen

- Onderzoekers ontdekken de belangrijkste oorzaak van energieverlies in spintronische materialen

 Onderzoekers vinden wereldwijd hints van Israëlische spyware
Onderzoekers vinden wereldwijd hints van Israëlische spyware- Hoe gebruik je een 9-volt batterij om LEDs te voeden
- Hoe fotonen per seconde te berekenen
- Hoe wetenschappers nieuwe kleuren uitvinden
- Bij longitudinaal onderzoek gedroogde bloedvlekmonsters spelen een rol
- Onderzoekers voeren eerste natuurkundige basissimulatie uit van de impact van gerecyclede atomen op plasmaturbulentie
- Wetenschappers ontwikkelen nieuwe meta-oppervlakken voor diepe UV-beeldvorming
- Twitter wint momentum met tweede winstgevende kwartaal op rij (update)
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
