Wetenschap
Machine learning versnelt simulaties in materiaalkunde
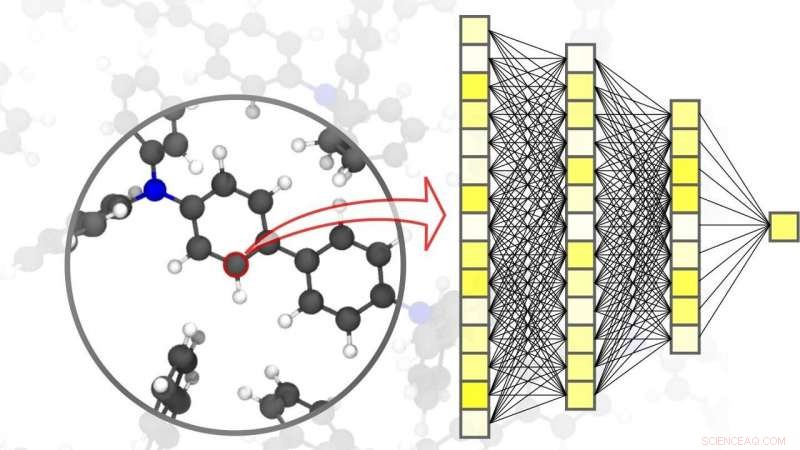
Neurale netwerken maken nauwkeurige simulaties in de materiaalwetenschap mogelijk - tot op het niveau van individuele atomen. Krediet:Pascal Friederich, KIT
Onderzoek, ontwikkeling, en de productie van nieuwe materialen zijn sterk afhankelijk van de beschikbaarheid van snelle en tegelijkertijd nauwkeurige simulatiemethoden. machinaal leren, waarin kunstmatige intelligentie (AI) autonoom nieuwe kennis verwerft en toepast, zullen onderzoekers straks in staat stellen complexe materiële systemen te ontwikkelen in een puur virtuele omgeving. Hoe werkt dit, en welke toepassingen zullen hiervan profiteren? In een artikel gepubliceerd in de Natuurmaterialen logboek, een onderzoeker van het Karlsruhe Institute of Technology (KIT) en zijn collega's uit Göttingen en Toronto leggen het allemaal uit.
Digitalisering en virtualisatie worden steeds belangrijker in een breed scala van wetenschappelijke disciplines. Een van deze disciplines is materiaalkunde:onderzoek, ontwikkeling, en de productie van nieuwe materialen zijn sterk afhankelijk van de beschikbaarheid van snelle en tegelijkertijd nauwkeurige simulatiemethoden. Dit, beurtelings, is gunstig voor een breed scala aan verschillende toepassingen - van efficiënte energieopslagsystemen, zoals die welke onmisbaar zijn voor het gebruik van hernieuwbare energiebronnen, naar nieuwe medicijnen, voor wiens ontwikkeling inzicht in complexe biologische processen vereist is. AI- en machine learning-methoden kunnen simulaties in materiaalwetenschappen naar een hoger niveau tillen. "Vergeleken met conventionele simulatiemethoden op basis van klassieke of kwantummechanische berekeningen, het gebruik van neurale netwerken die specifiek zijn afgestemd op materiaalsimulaties stelt ons in staat een aanzienlijk snelheidsvoordeel te behalen, " legt natuurkundige en AI-expert professor Pascal Friederich uit, Hoofd van de AiMat-Artificial Intelligence for Materials Sciences onderzoeksgroep bij KIT's Institute of Theoretical Informatics (ITI). "Met snellere simulatiesystemen, wetenschappers zullen grotere en complexere materiële systemen kunnen ontwikkelen in een puur virtuele omgeving, en om ze te begrijpen en te optimaliseren tot op atomair niveau."
Hoge precisie van het atoom tot het materiaal
In een artikel gepubliceerd in Natuurmaterialen , Pascal Friederich, die ook associate group leader is van de Nanomaterials by Information-Guided Design-divisie bij KIT's Institute of Nanotechnology (INT), presenteert, samen met onderzoekers van de Universiteit van Göttingen en de Universiteit van Toronto, een overzicht van de basisprincipes van machine learning die worden gebruikt voor simulaties in materiaalwetenschappen. Dit omvat ook het data-acquisitieproces en actieve leermethoden. Machine learning-algoritmen stellen niet alleen kunstmatige intelligentie in staat om de invoergegevens te verwerken, maar ook om patronen en correlaties te vinden in grote datasets, leer van ze, en autonoom voorspellingen en beslissingen te nemen. Voor simulaties in materiaalkunde, het is belangrijk om een hoge nauwkeurigheid te bereiken over verschillende tijd- en grootteschalen, variërend van het atoom tot het materiaal, terwijl de rekenkosten worden beperkt. In hun artikel, de wetenschappers bespreken ook verschillende huidige toepassingen, zoals kleine organische moleculen en grote biomoleculen, structureel ongeordend vast, vloeistof, en gasvormige materialen, evenals complexe kristallijne systemen, bijvoorbeeld metaal-organische raamwerken die kunnen worden gebruikt voor gasopslag of voor scheiding, voor sensoren of voor katalysatoren.
Nog meer snelheid met hybride methoden
Om de mogelijkheden van materiaalsimulaties in de toekomst verder uit te breiden, de onderzoekers uit Karlsruhe, Göttingen, en Toronto suggereren de ontwikkeling van hybride methoden:deze combineren machine learning (ML) en moleculaire mechanica (MM) methoden. MM-simulaties gebruiken zogenaamde krachtvelden om de krachten te berekenen die op elk afzonderlijk deeltje inwerken en zo bewegingen te voorspellen. Aangezien de mogelijkheden van de ML- en MM-methoden vrij gelijkaardig zijn, een nauwe integratie met variabele overgangsgebieden is mogelijk. Deze hybride methoden kunnen in de toekomst de simulatie van grote biomoleculen of enzymatische reacties aanzienlijk versnellen, bijvoorbeeld.
 NOAA's GOES-16 klaar om voorspellingen nog meer te verbeteren
NOAA's GOES-16 klaar om voorspellingen nog meer te verbeteren- Lijst met insecten die kunnen overwinteren
- Modellering van vroegere en toekomstige gletsjeroverstromingen in het noorden van Groenland
- Door droogte veroorzaakte veranderingen in bossamenstelling versterken de effecten van klimaatverandering
- NASA-NOAA-satelliet vindt een zwakkere, overgang van tropische storm Halong
Hoofdlijnen
- Karkas van Noord-Atlantische rechtse walvis gespot in Massachusetts
- Noordelijke maïsbladziekte-genen geïdentificeerd in nieuwe studie
- Vogelherkenning
- Wanneer is lachen een medisch symptoom?
- Hoe beïnvloedt DNA-replicatie je lichaam?
- Wat zijn de functies van glazen schuif- en afdekglaasjes?
- UFO-psychologie
- Biologen ontdekken tastzin van bacteriën
- Prozac in oceaanwater een mogelijke bedreiging voor het zeeleven, studie vondsten
- Complex leven is ontstaan uit de toevallige koppeling van kleine moleculen

- Hoe vindt u hoeveel mol in een verbinding

- Beoordeling van het begin van calciumfosfaatkiemvorming door gehyperpolariseerde real-time NMR

- Wetenschappers bakken glutenvrij brood met een revolutionaire technologie

- Wetenschappers ontwikkelen lithium-ionbatterij die niet vlam vat


 Miljoenen snikheet in het westen van de VS terwijl Canada noodmaatregelen neemt
Miljoenen snikheet in het westen van de VS terwijl Canada noodmaatregelen neemt- Hubble Hubble:telescoop levert verbluffende nieuwe beelden van twee planetaire nevels
- Indonesië geeft ontwerp 737 MAX de schuld van Lion Air-crash:rapport
- Exotische legeringen voor potentiële energietoepassingen
- Nagelbed borstimplantaat schrikt kankercellen af
- Niches in Deserts
- Kunnen computers helpen om partijdige scheidslijnen te dichten?
- Kwantuminformatie-uitwisseling over lange afstand - succes op nanoschaal
- Elektronica
- Biologie
- Zonsverduistering
- Wiskunde
- French | Italian | Spanish | Portuguese | Swedish | German | Dutch | Danish | Norway |

-
Wetenschap © https://nl.scienceaq.com
